
Charting a Path to Success in Virtual Screening

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

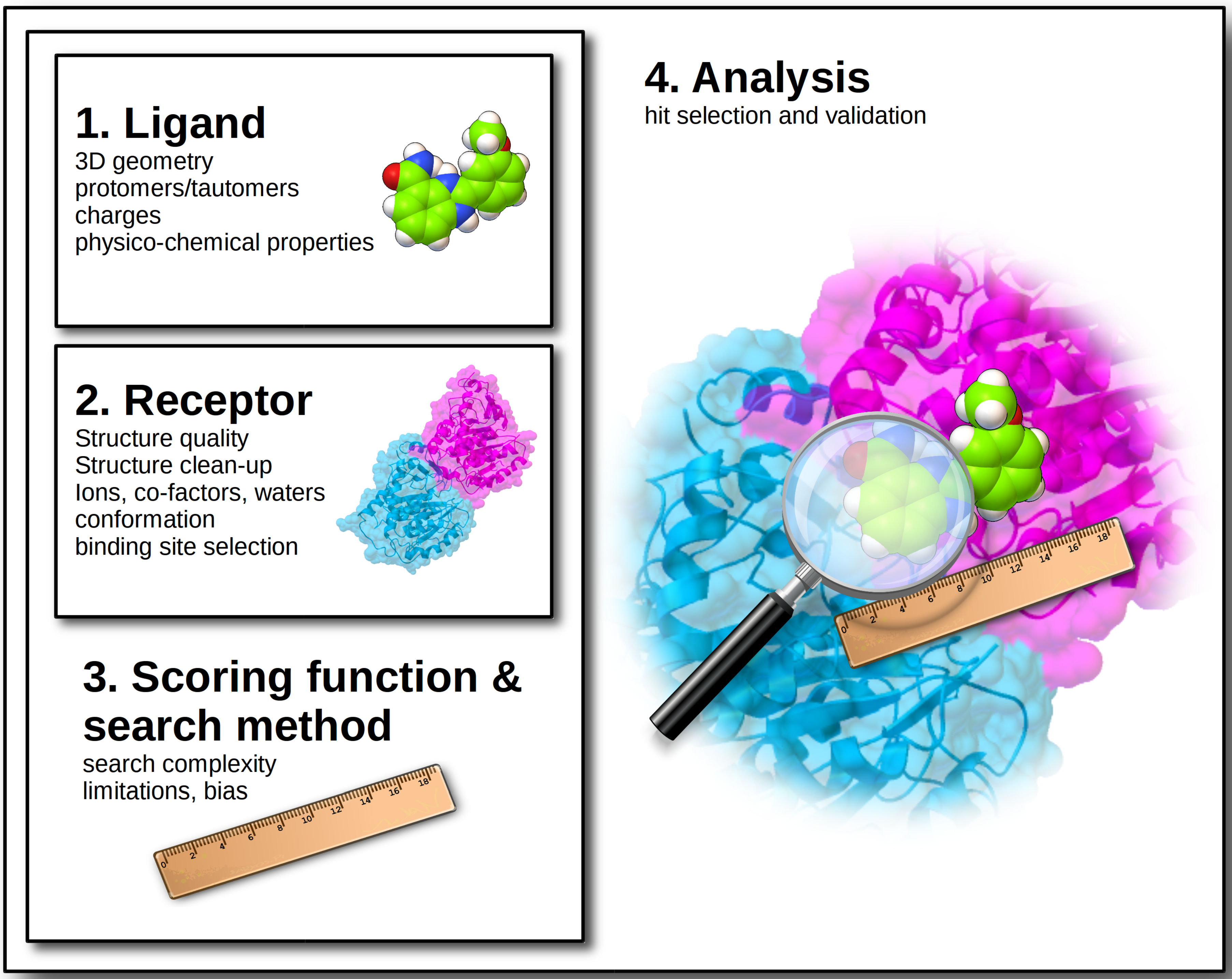
2. Ligand Structures
2.1. Accurate 3D Geometries
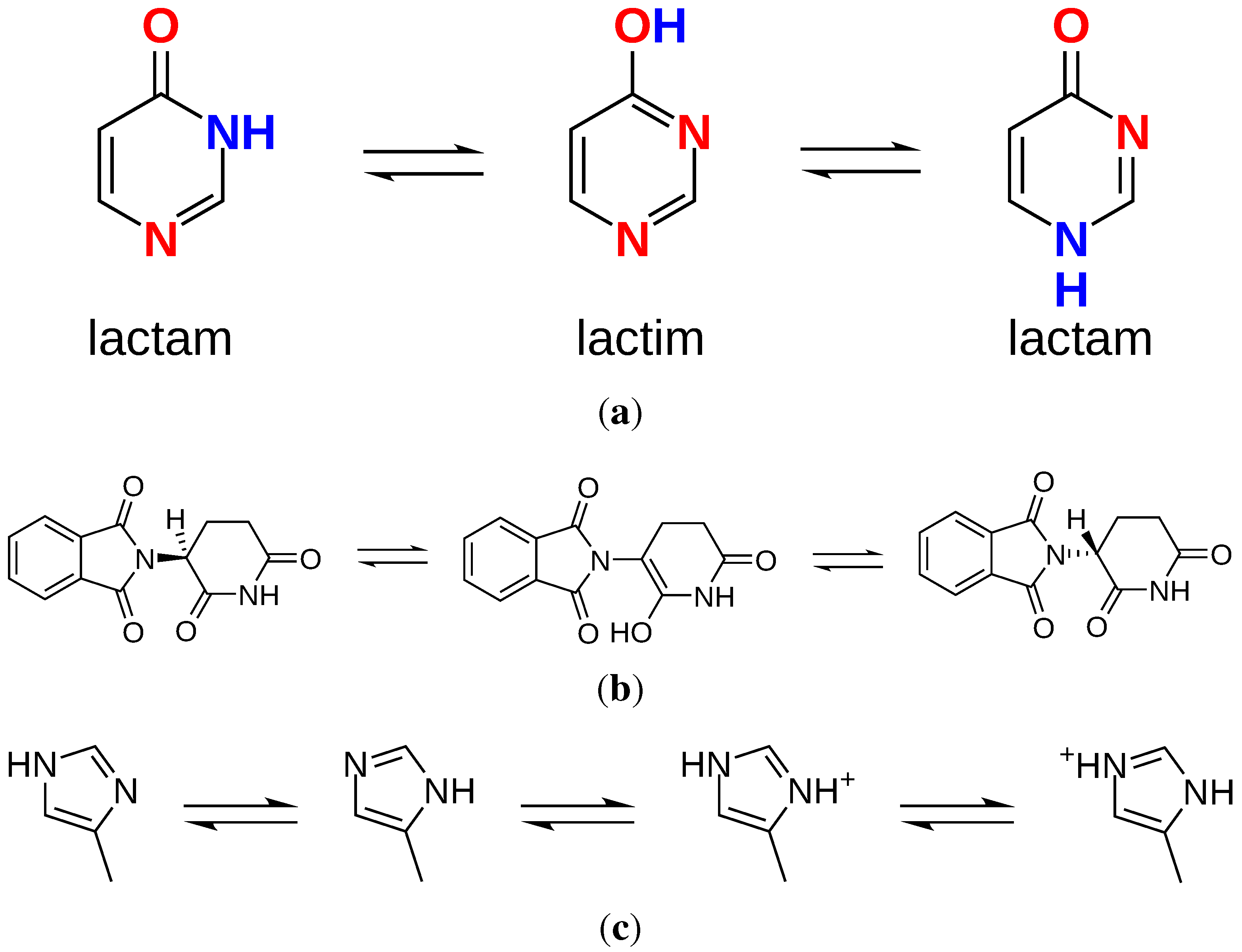
2.2. Tautomers and Protonation States


2.3. Charges
2.4. Physicochemical Properties
3. Target Structure
3.1. Structure Quality
3.2. Structure Clean-Up

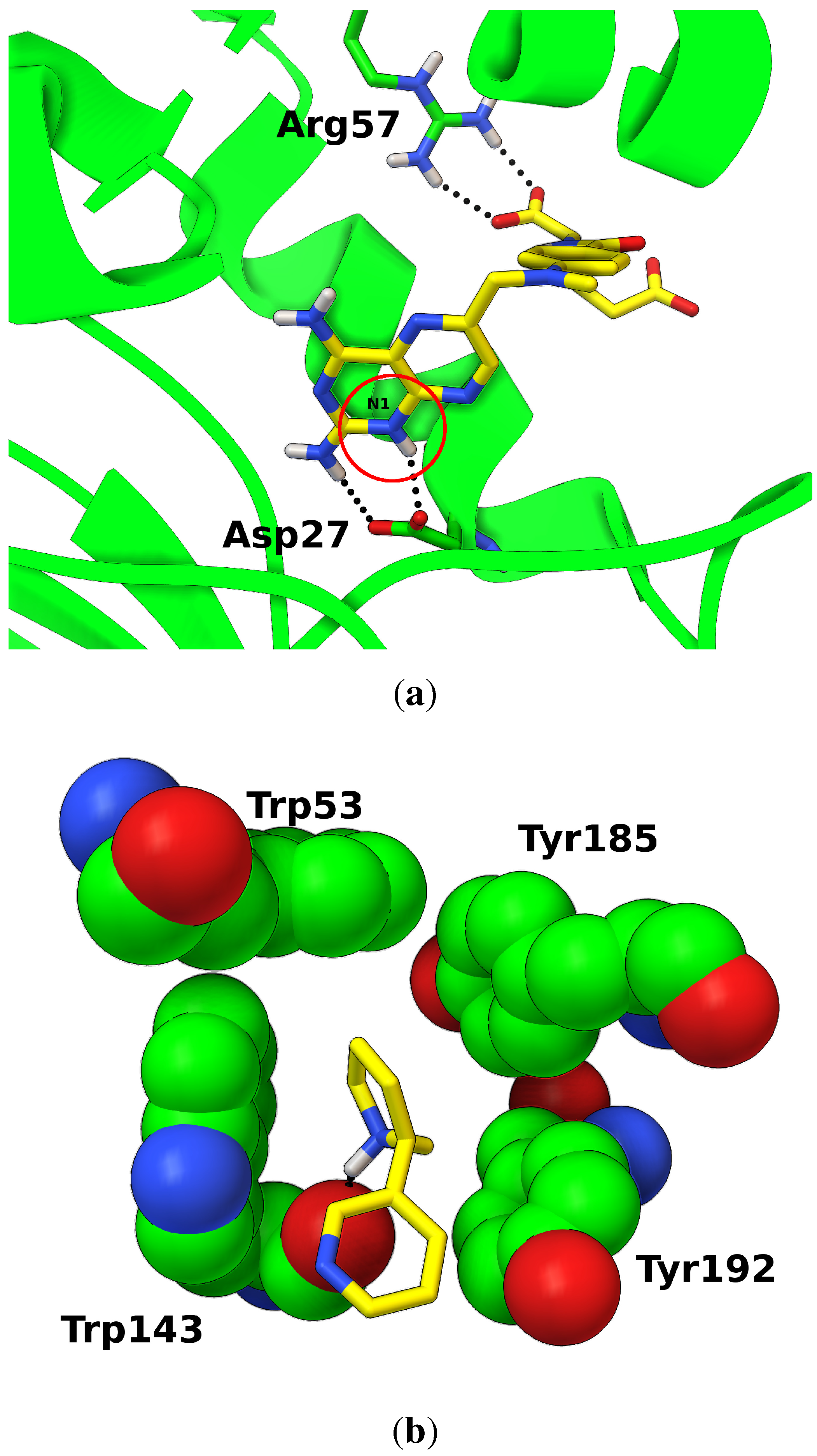
3.3. Protonation States
3.4. Coordinating Metal Ions, Co-Factors and Waters
3.5. Structure Conformation

3.6. Binding Site Selection
4. Scoring Function and Search Method
5. Results and Assay
5.1. Hit Selection
5.2. Hit Validation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walters, W.P.; Namchuk, M. Designing screens: How to make your hits a hit. Nat. Rev. Drug Discov. 2003, 2, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Perola, E.; Xu, K.; Kollmeyer, T.M.; Kaufmann, S.H.; Prendergast, F.G.; Pang, Y.P. Successful virtual screening of a chemical database for farnesyltransferase inhibitor leads. J. Med. Chem. 2000, 43, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Grüneberg, S.; Stubbs, M.T.; Klebe, G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: Strategy and experimental confirmation. J. Med. Chem. 2002, 45, 3588–3602. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Klabunde, T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the alpha1A adrenergic receptor. J. Med. Chem. 2005, 48, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Ferreira, R.S.; Irwin, J.J.; Shoichet, B.K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 20, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Eidam, O.; Fish, I.; Hollander, J.; Figaroa, F.; Nachane, R.; Irwin, J.J.; Shoichet, B.K.; Siegal, G. Increasing Chemical Space Coverage by Combining Empirical and Computational Fragment Screens. ACS Chem. Biol. 2014, 9, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Al Olaby, R.R.; Cocquerel, L.; Zemla, A.; Saas, L.; Dubuisson, J.; Vielmetter, J.; Marcotrigiano, J.; Khan, A.G.; Catalan, F.V.; Perryman, A.L.; et al. Identification of a novel drug lead that inhibits HCV infection and cell-to-cell transmission by targeting the HCV E2 glycoprotein. PLoS ONE 2014, 9, e111333. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Yu, W.; Wang, X.; Ekins, S.; Forli, S.; Li, S.G.; Freundlich, J.S.; Tonge, P.J.; Olson, A.J. A Virtual Screen Discovers Novel, Fragment-Sized Inhibitors of Mycobacterium tuberculosis InhA. J. Chem. Inf. Model. 2015, 55, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Babbage, C. Passages from the Life of a Philosopher; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; Mosi, R.; Kalk, K.H.; van der Veen, B.A.; Dijkhuizen, L.; Withers, S.G.; Dijkstra, B.W. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat. Struct. Mol. Biol. 1999, 6, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules 2015, 20, 13997–14021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.L.; Tan, E.; et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model. 2013, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Tebben, A.J.; Cheney, D.L.; Shelley, J.C. Macrocycle conformational sampling with MacroModel. J. Chem. Inf. Model. 2014, 54, 2680–2696. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Botta, M. Lennard-Jones potential and dummy atom settings to overcome the AutoDock limitation in treating flexible ring systems. J. Chem. Inf. Model. 2007, 47, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics, 2013. Available online: http://www.rdkit.org (accessed on 14 July 2015).
- ChemAxon. Standardizer, JChem 15.7.13.0, 2015. Available online: http://www.chemaxon.com (accessed on 14 July 2015).
- Miteva, M.A.; Guyon, F.; Tufféry, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef] [PubMed]
- CACTVS-Server. Online SMILES Translator and Structure File Generator, 2010. Available online: http://cactus.nci.nih.gov/translate/ (accessed on 20 July 2015).
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely available conformer generation methods: How good are they? J. Chem. Inf. Model. 2012, 52, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, P.M.; Vignesh, N.; Kumar, G.R.; Doble, M. Computational approaches to enhance activity of taxanes as antimitotic agent. Med. Chem. Res. 2012, 21, 2557–2570. [Google Scholar] [CrossRef]
- Sayle, R.A. So you think you understand tautomerism? J. Comput.-Aided Mol. Des. 2010, 24, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sitzmann, M.; Ihlenfeldt, W.D.; Nicklaus, M.C. Tautomerism in large databases. J. Comput.-Aided Mol. Des. 2010, 24, 521–551. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; John Wiley & Sons: New York, NY, USA, 2007. [Google Scholar]
- Martin, Y.C. Let’s not forget tautomers. J. Comput.-Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; **. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble docking into multiple crystallographically derived protein structures: An evaluation based on the statistical analysis of enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Kovacs, J.A.; Abagyan, R.A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 2005, 127, 9632–9640. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Sanschagrin, P.C.; Greenwood, J.R.; Repasky, M.P.; Sherman, W.; Farid, R. Improving database enrichment through ensemble docking. J. Comput.-Aided Mol. Des. 2008, 22, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.T.; Liu, J.; Estiu, G.; Oltvai, Z.; Wiest, O. Identification of novel bacterial histidine biosynthesis inhibitors using docking, ensemble rescoring, and whole-cell assays. Bioorg. Med. Chem. 2010, 18, 5148–5156. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- IBM. World Community Grid, 2007. Available online: http://www.worldcommunitygrid.org/about_us/viewAboutUs.do (accessed on 20 July 2015).
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Davis, I.W.; Raha, K.; Head, M.S.; Baker, D. Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci. 2009, 18, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins: Struct. Funct. Bioinform. 2009, 74, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Sanchez, R. Beyond structural genomics: Computational approaches for the identification of ligand binding sites in protein structures. J. Struct. Funct. Genomics 2011, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Westhof, E.; Hanessian, S. Docking of aminoglycosides to hydrated and flexible RNA. J. Med. Chem. 2006, 49, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Perola, E.; Walters, W.P.; Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins Struct. Funct. Bioinform. 2004, 56, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.A.; Morris, G.M.; Biggin, P.C. One size does not fit all: The limits of structure-based models in drug discovery. J. Chem. Theory Comput. 2013, 9, 4266–4274. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Gasch, T.; Stahl, M. Scoring functions for protein–ligand interactions: A critical perspective. Drug Discov. Today Technol. 2004, 1, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Chia-en, A.C.; Chen, W.; Gilson, M.K. Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. USA 2007, 104, 1534–1539. [Google Scholar]
- Merlitz, H.; Wenzel, W. Comparison of stochastic optimization methods for receptor–ligand docking. Chem. Phys. Lett. 2002, 362, 271–277. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, X.; Li, Y.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model. 2009, 49, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.A.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar] [CrossRef]
- Balius, T.E.; Mukherjee, S.; Rizzo, R.C. Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons. J. Comput. Chem. 2011, 32, 2273–2289. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Santiago, D.N.; Forli, S.; Santos-Martins, D.; Olson, A.J. Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: Participation in the SAMPL4 protein–ligand binding challenge. J. Comput.-Aided Mol. Des. 2014, 28, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2007, 41, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łażniewski, M.; Grotthuss, M.V.; Rychlewski, L.; Ginalski, K. VoteDock: Consensus docking method for prediction of protein–ligand interactions. J. Comput. Chem. 2011, 32, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Gleeson, D. QM/MM as a tool in fragment based drug discovery. A cross-docking, rescoring study of kinase inhibitors. J. Chem. Inf. Model. 2009, 49, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Gallicchio, E.; Deng, N.; He, P.; Wickstrom, L.; Perryman, A.L.; Santiago, D.N.; Forli, S.; Olson, A.J.; Levy, R.M. Virtual screening of integrase inhibitors by large scale binding free energy calculations: The SAMPL4 challenge. J. Comput.-Aided Mol. Des. 2014, 28, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Slynko, I.; Scharfe, M.; Rumpf, T.; Eib, J.; Metzger, E.; Schüle, R.; Jung, M.; Sippl, W. Virtual screening of PRK1 inhibitors: Ensemble docking, rescoring using binding free energy calculation and QSAR model development. J. Chem. Inf. Model. 2014, 54, 138–150. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Pospisil, P.; Pos, W.; Folkers, G.; Vermeulen, N.P. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J. Med. Chem. 2005, 48, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Forli, S.; He, P.; Perryman, A.; Wickstrom, L.; Vijayan, R.; Tiefenbrunn, T.; Stout, D.; Gallicchio, E.; Olson, A.J.; et al. Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J. Phys. Chem. B 2014, 119, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Zartler, E.R. Quantum Tessera Consulting, 2013. Available online: http://www.quantumtessera.com/your-computation-is-only-as-good-as-your-experimental-follow-up/ (accessed on 14 July 2015).
- Lo, M.C.; Aulabaugh, A.; **, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lea, W.A.; Simeonov, A. Fluorescence polarization assays in small molecule screening. Expert Opin. Drug Discov. 2011, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.M.; Van Molle, I.; Baud, M.G.; Galdeano, C.; Geraldes, C.F.; Ciulli, A. Is NMR fragment screening fine-tuned to assess druggability of protein–protein interactions? ACS Med. Chem. Lett. 2013, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wielens, J.; Headey, S.J.; Rhodes, D.I.; Mulder, R.J.; Dolezal, O.; Deadman, J.J.; Newman, J.; Chalmers, D.K.; Parker, M.W.; Peat, T.S.; et al. Parallel Screening of Low Molecular Weight Fragment Libraries Do Differences in Methodology Affect Hit Identification? J. Biomol. Screen. 2013, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732-18758. https://doi.org/10.3390/molecules201018732
Forli S. Charting a Path to Success in Virtual Screening. Molecules. 2015; 20(10):18732-18758. https://doi.org/10.3390/molecules201018732
Chicago/Turabian StyleForli, Stefano. 2015. "Charting a Path to Success in Virtual Screening" Molecules 20, no. 10: 18732-18758. https://doi.org/10.3390/molecules201018732