1. Introduction
In an era facing the depletion of fossil fuels and the increasing environmental concerns related to their burning, much of the current efforts have been directed to the challenging search for more sustainable sources that can ensure continuous manufacture of those commodities that have improved the quality of human life. As an example, recent years have witnessed a rapid increase in the production of bio-based plastics and polymers, entirely derived from renewable sources, such as starch, cellulose, sugars, etc. [
1,
2]. In this context, vegetable oils, such as ricinoleic, linseed, soybean oils, etc., have also been regarded as convenient source of renewable feedstocks to be employed in the development of bio-based polyurethanes (PU) [
3,
4,
5,
6,
7,
8,
9].
Bio-based polyurethanes are available in a wide range of hardnesses, as well as in several formulations, to obtain different materials that allow energy savings, protect the environment, enhance safety, lead to innovative building systems, as well as several polyurethanes that have been studied for their preparation of composites [
1,
10,
11,
12,
13].
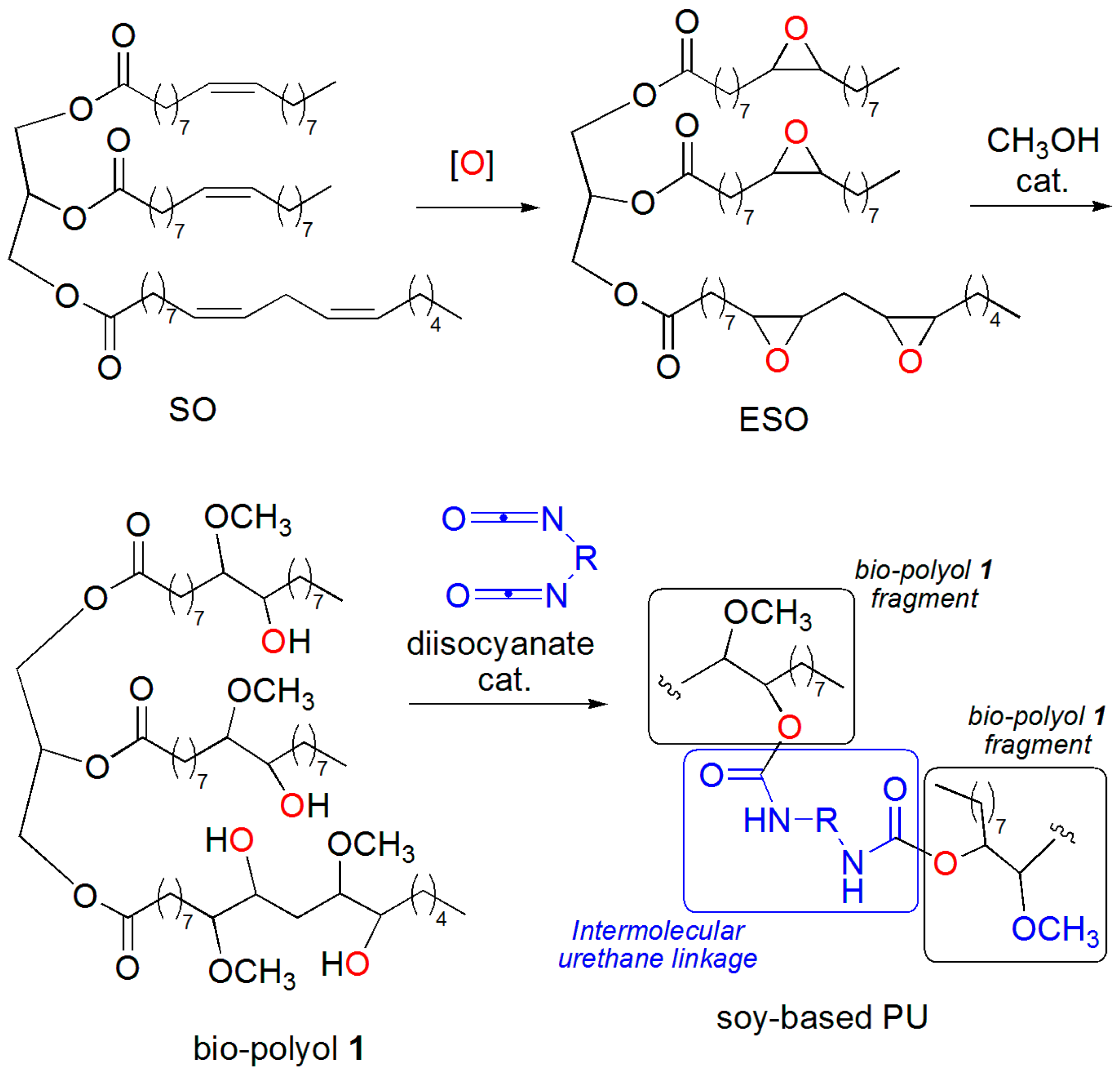
A practical approach to PU from vegetable oils involves epoxidation of the carbon-carbon double bonds of unsaturated fatty ester moieties and subsequent epoxide ring-opening reaction by nucleophilic reagents, providing the hydroxyl functionalities that form the urethane network upon reaction with isocyanates [
3]. This is exemplified by the conversion of soybean oil (SO) into soy-based PU, through preliminary epoxidation, followed by methanolysis of the epoxidized oil (ESO), and reaction of the ensuing soy-methanol polyol 1 with suitable diisocyanates (
Scheme 1).
In this context, special attention has been paid to oxidation methods employing organocatalysts [
14,
15] or based on mild and safe oxidants, such as H
2O
2 [
16,
17] organic (hydro)peroxides [
18,
19,
20] and even O
2 [
21]. On the other hand, typical protocols prescribe the use of strong Brønsted acids (e.g.,
p-Toluene sulfonic acid SA, H
3PO
4, and HBF
4) to catalyze the epoxide ring-opening reaction step, while hazardous Lewis acids (e.g., tin-, mercury-compounds) and/or toxic bases (e.g., tertiary amines) are usually employed in the formation of urethane linkages [
22,
23,
24].
In our ongoing efforts aimed at searching new green catalytic methods [
25,
26,
27,
28] our interest in the area is driven by the search for an efficient, cost-effective, and environmentally safer conversion of epoxidized soybean oil (ESO) into bio-polyol 1 and then into soy-based PU (
Scheme 1), with the ultimate goal of moving the whole process from laboratory bench to a pilot scale and then on to a manufacturing scale. Bio-polyol 1 shows interesting physico-chemical properties that makes it an excellent substitute for petrochemical polyols in the manufacture of some soy-based PUs.
In recent years, polyurethanes have been prepared by reacting a soybean oil-based polyol with different isocyanates to give foams, elastomers, coatings, and adhesives [
1,
8] in addition, the influence of a catalysts on gelling and blowing reactions was studied to compare the properties of polymers [
22,
23,
24].
By taking advantage of the high versatility of dichlorodioxo-molybdenum(VI), MoCl
2O
2, as well as Lewis acid catalyst [
29], this work describes our efforts in applying MoCl
2O
2 to the direct conversion of epoxidized soybean oil (ESO) into PU in a one-pot reaction that encompasses both the epoxide ring-opening stage and the subsequent urethane linkage formation step. In this way, the additional steps of acid catalyst neutralization and removal of the ensuing salts are completely bypassed. This, coupled with high efficiency and better environmental acceptance of MoCl
2O
2 over other metal-based catalysts, makes the whole process amenable to industrial scale-up.
2. Results and Discussion
Molybdenum is much less toxic than typical heavy metals. For this reason, molybdenum-based compounds have found several applications as clean alternatives to heavy metals in the petroleum and plastic industries. In particular, MoCl
2O
2, and its related complexes, serve well as Lewis acid catalysts for several organic transformations [
29], ranging from oxidation to acylation, reduction, and carbamylation [
30,
31,
32]
Reportedly, MoCl
2O
2 can efficiently catalyze the methanolysis of a range of terminal epoxides to β-methoxy alcohols, for which 5 mol % of catalyst generally suffices to drive reactions to completion within 1–5 h at 50 °C or, in some cases, even at room temperature [
33].
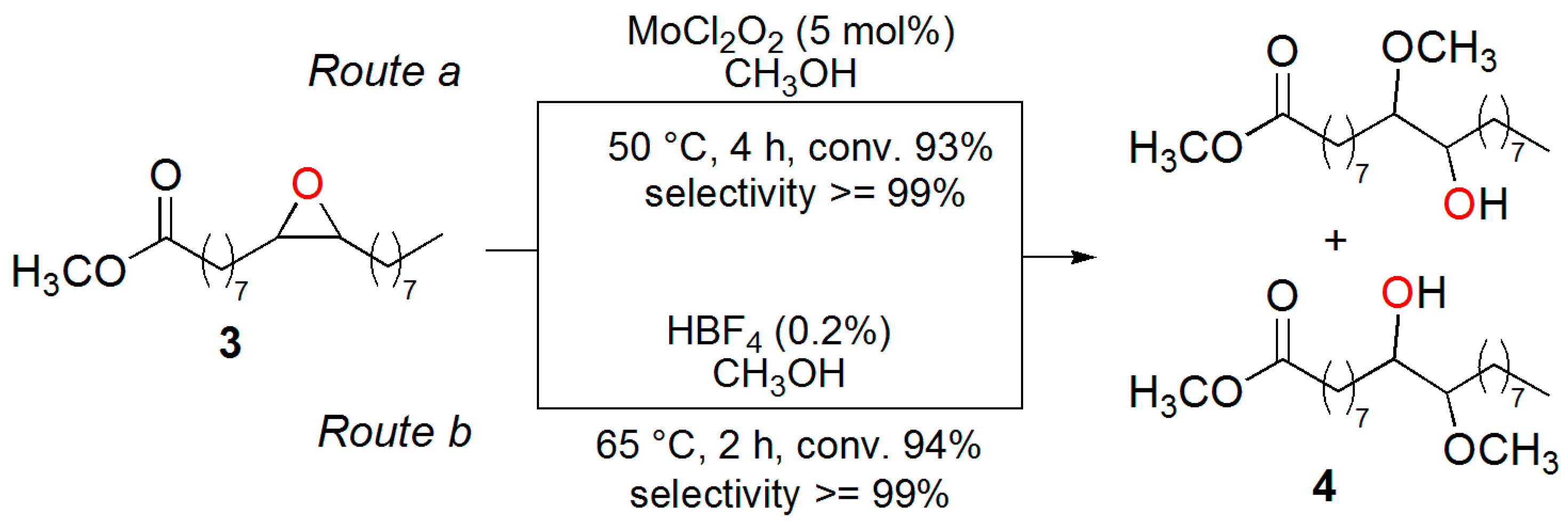
In order to test whether this protocol could be extended to the methanolysis of such internal epoxides as those of ESO (
Scheme 1), we deemed it useful to first study the MoCl
2O
2-catalyzed epoxide-ring opening reaction of methyl oleate epoxide (
3) with methanol under the reported conditions. To this purpose, methyl oleate epoxide (
3) was prepared by reaction of commercial methyl oleate (
2) with H
2O
2/formic acid, according to a widely-exploited industrial protocol [
17]. Thus, upon treatment of epoxide
3 with 5 mol % MoCl
2O
2 in CH
3OH at 50 °C (
Scheme 2, route a), GC-MS analysis of the reaction mixture revealed that substrate conversion reached 93% after 4 h. Additionally, the MS and spectral data of the crude product displayed the characteristic features of methyl 10(9)-hydroxy-9(10)-methoxy-octadecanoate
4, which was obtained with selectivity ≥99% as a roughly equimolar mixture of regioisomers. For unambiguous identification of β-methoxy alcohol
4, a reference sample was made available upon subjecting methyl oleate epoxide (
3) to conventional methanolysis by tetrafluoroboric acid (HBF
4) catalysis, according to route b of
Scheme 2 [
34,
35,
36].
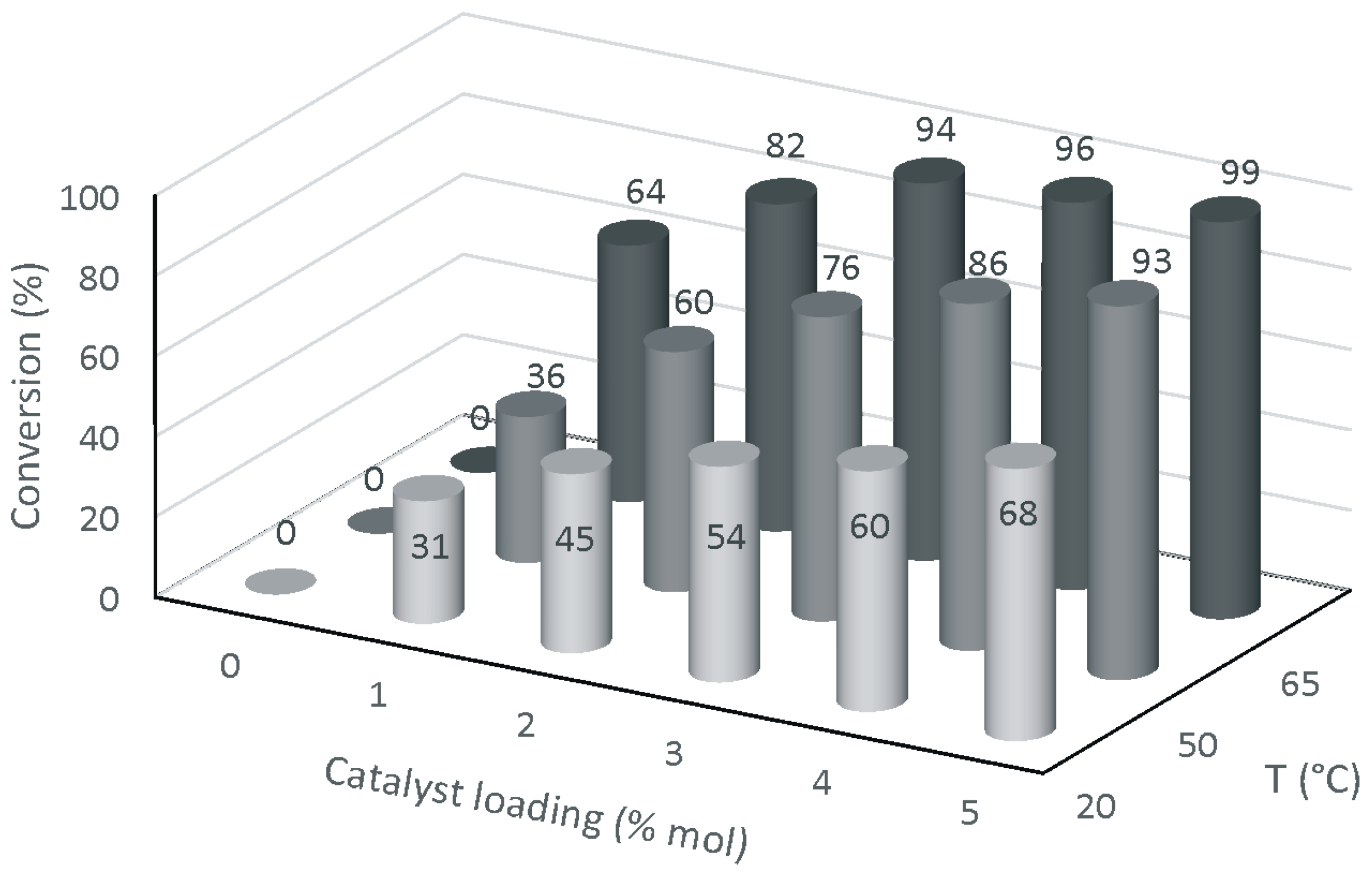
Next, we turned to check whether the reaction conditions adopted were optimal for the case at hand. Thus, the reaction was studied under variable conditions of catalyst loading (1–5 mol %) and temperature (20–65 °C) at the fixed reaction time of 4 h. Results are collected in the diagram of
Figure 1.
As shown in
Figure 1, substrate conversion is directly correlated to catalyst loading and temperature. The GC-MS analysis of the reaction mixtures revealed that, in the range of temperatures and catalyst loading explored, product selectivity was retained, being always higher than 99%. Overall, these results point out that an increase in temperature can compensate for the decrease of catalyst loading, without significantly affecting product selectivity. This means that as long as the temperature is increased, the amount of catalyst can be reduced, which is often the most desirable condition for the development of economical processes. For instance, using 3 mol % of MoCl
2O
2 at 65 °C allows reaction to reach 94% conversion after 4 h (
Figure 1), which is the same result as when the methanolysis is carried out under typical conditions (
Scheme 2, route b).
In light of these results, MoCl
2O
2 appears well-suited to be employed in the methanolysis of epoxidized fatty esters, as an alternative to HBF
4. Therefore, we turned to explore its catalytic performance in the epoxide ring-opening reaction of ESO by methanol. Instructed by the new experimental conditions found for the methanolysis of methyl oleate epoxide (
Figure 1), we performed all experiments at 65 °C, while catalyst loading was varied from 1 up to 3 mol %.
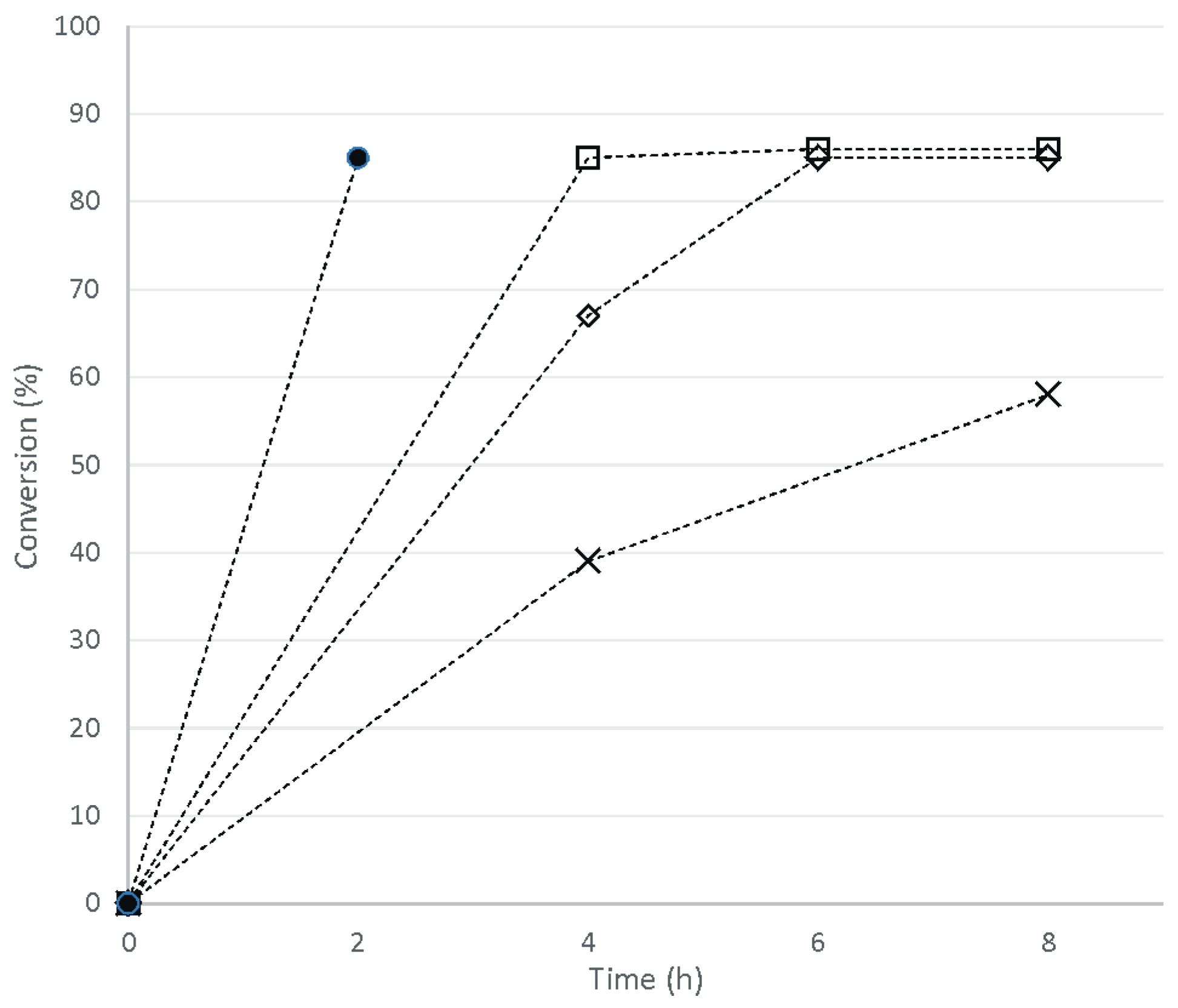
1H-NMR analysis of reaction mixtures was preferred over GC-MS techniques, allowing us to estimate substrate conversion and product identity, using the reference spectral data of bio-polyol
1 as obtained from typical HBF
4-catalyzed methanolysis of ESO at 65 °C for 2 h [
36,
37]. Shown in
Figure 2, methanolysis of ESO catalyzed by HBF
4 attains a maximum of substrate conversion of 85% at 2 h, as estimated based on signal integration of residual epoxide protons (3.15–2.74 ppm) in the
1H-NMR spectrum of bio-polyol
1 (
Figure 3b) in comparison with the corresponding signal integrations in the spectrum of ESO (
Figure 3a). The singlet at 3.65 ppm in the spectrum of
Figure 3b can be assigned to the resonance of methoxyl protons of fatty acid methyl esters (~7%), suggesting that transesterification is concomitant to the epoxide ring-opening reaction. Yet, it is seen that longer reaction times cause transesterification of triglycerides to take place at a rate of approximately 2% of fatty acid methyl esters formed per each additional hour of reaction.
Figure 2 shows that, in the MoCl
2O
2-catalyzed methanolysis of ESO at 65 °C, the maximum substrate conversion achievable is still as high as 85%–86%, regardless of catalyst loading and reaction time. However, the use of 1 mol % of MoCl
2O
2 requires as long as 24 h to allow conversion to reach 80%, while on doubling or tripling the catalyst concentration, 85% conversion can be smoothly reached within 4 or 6 h, respectively.
In all of the cases examined, reactions led to the selective conversion of ESO into bio-polyol
1, as one can judge from the comparison of
1H-NMR spectral profiles of
1 obtained by HBF
4 (
Figure 3b) or MoCl
2O
2 (2 mol %,
Figure 3c). In the
1H-NMR spectrum of
Figure 3c, the signal at 3.65 ppm indicates the presence of minor amounts (~3%) of methyl ester side products. Unlike the case with HBF
4 catalyst, their amount was found to not increase appreciably for prolonged reaction times, consistent with the weak propensity of MoCl
2O
2 to catalyze transesterification reactions [
31,
38].
Other hints of the conversion of ESO into bio-polyol
1 in the reaction with CH
3OH/MoCl
2O
2 come from the measurement of physicochemical properties, such as density, viscosity, and the average number of hydroxyl and epoxy functionalities (
Table 1).
Data reported in
Table 1 indicate that it is possible to obtain the bio-polyol
1 with 3.45 hydroxyl groups per molecule, instead of 4, as would expect based on the average number of epoxy groups. This discrepancy has already been explained as result of the presence of ethereal bonds due to concurrent intramolecular epoxide ring-opening (cross-linking process).
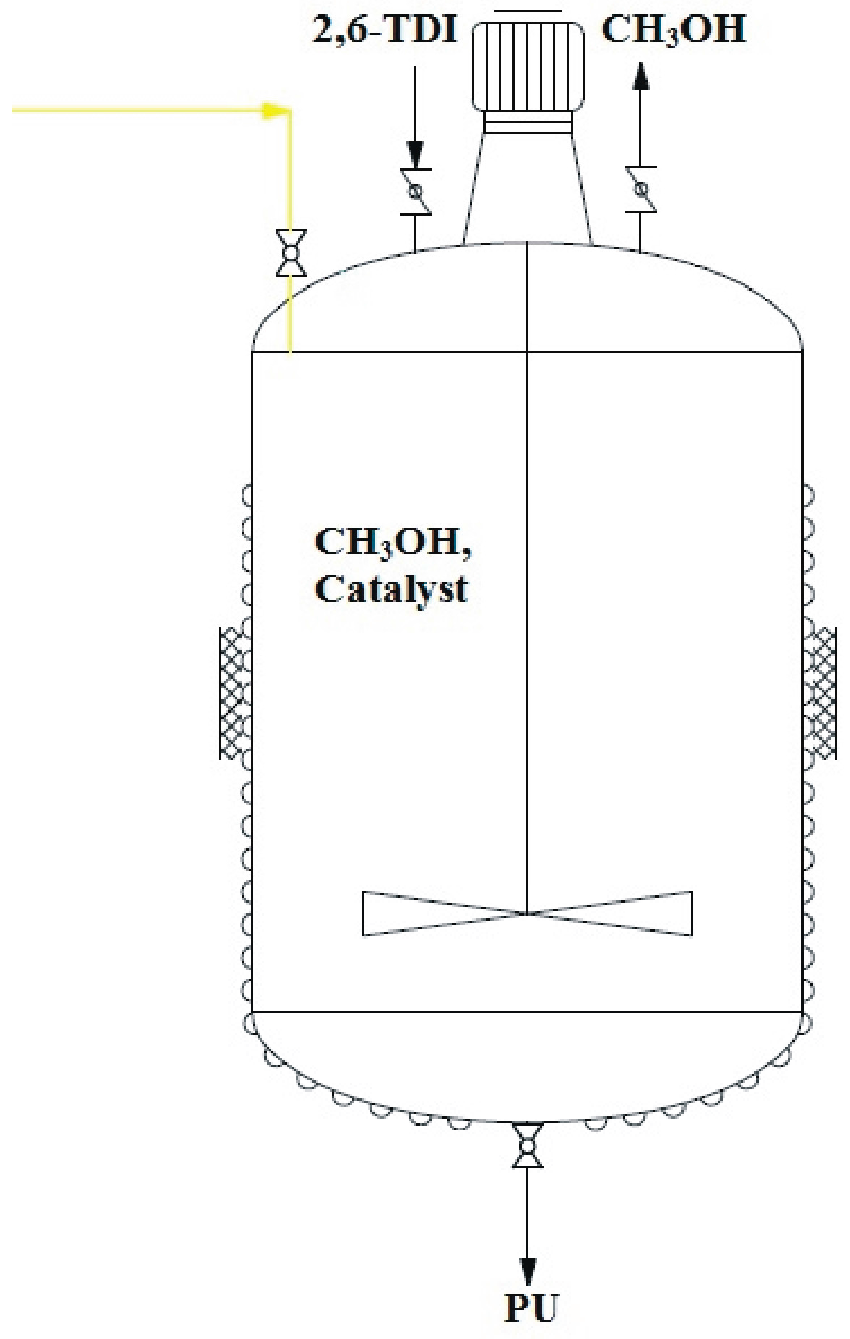
Next, we turned our attention to the “step 2” of the one-pot MoCl
2O
2-catalyzed soy-based polyurethane production, i.e., the reaction of bio-polyol
1 with toluene 2,6-diisocyanate (2,6-TDI) (
Scheme 3).
Brückner and coworkers have recently studied catalytic activity of MoCl
2O
2 and related complexes in the formation of mono-, di-, tri-, and tetracarbamates from a variety of alcohols and isocyanates [
30]. They found that these reactions proceed to completion at room temperature in few minutes using as little as 0.1–1 mol % of the catalyst in chlorinated solvents.
Polyurethanes were prepared according to a reported procedure in the experimental section, where each polyol obtained by methanolysis reaction with 1–5 mol % of catalyst on respect to epoxide, after evaporation in vacuo of methanol, was mixed in a plastic cup with 2,6-TDI for 30 s using a high-speed mixer. The gel times at room temperature for each compound were 4–12 min.
Polyurethanes obtained from the polyols were quite similar, which was reflected in their physical and mechanical properties.
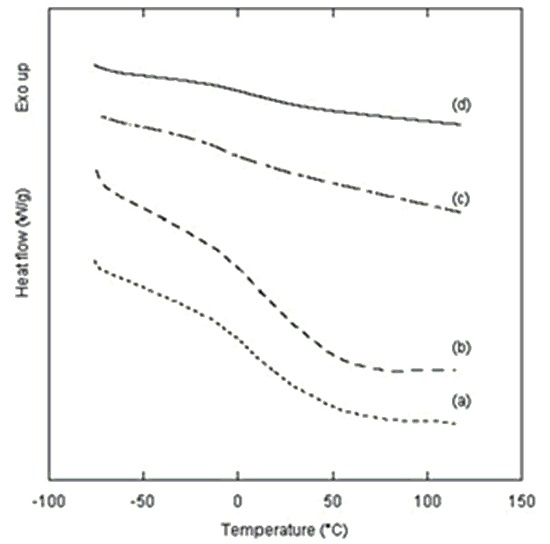
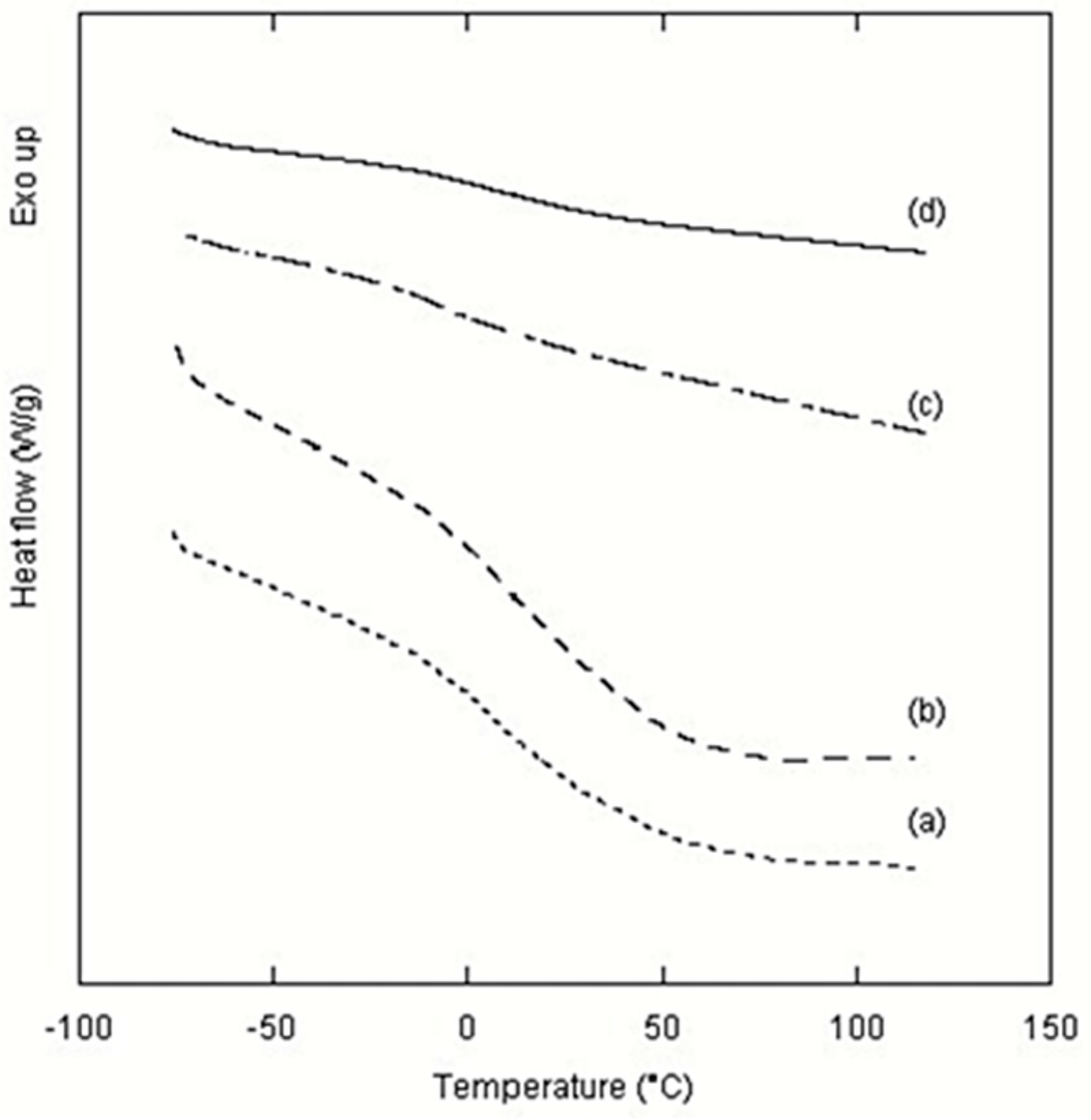
The thermal properties of polyurethane samples, obtained using different amounts of catalyst, were analyzed by DSC seven days after their synthesis (
Figure 4). No thermal evidence of melting, crystallization or decomposition were detected in the studied range of temperature from −80 °C to 120 °C. It turned out that all samples were in an amorphous state and thermally stable.
Thermal events attributable to glass transitions were observed only for samples prepared at lower catalyst quantity, 1 mol % and 2 mol %, with midpoints in the range between 9.4 and 10.5 °C (
Figure 4a,b). These glass transition temperatures (
Tg) indicate that polyurethane samples prepared using the one-pot synthesis herein reported with 1 mol % and 2 mol % of catalyst are rubbery at room temperature.
As a control experiment, we prepared the same bio-PU using the literature procedure [
36] based on the thermal catalysis. In this case, heating of the mixture for 24 h at 110 °C was necessary to complete the polymerization reaction, and a rubbery polymer with a
Tg of 20 °C was obtained.
Another piece of evidence supporting the suitability of the proposed method was obtained by polymerizing bio-polyol 1, achieved with the HBF4, in the presence of fresh 2 mol % of MoCl2O2. The PU thus obtained displayed a Tg value very close to that of the polymer achieved using the one-pot method.
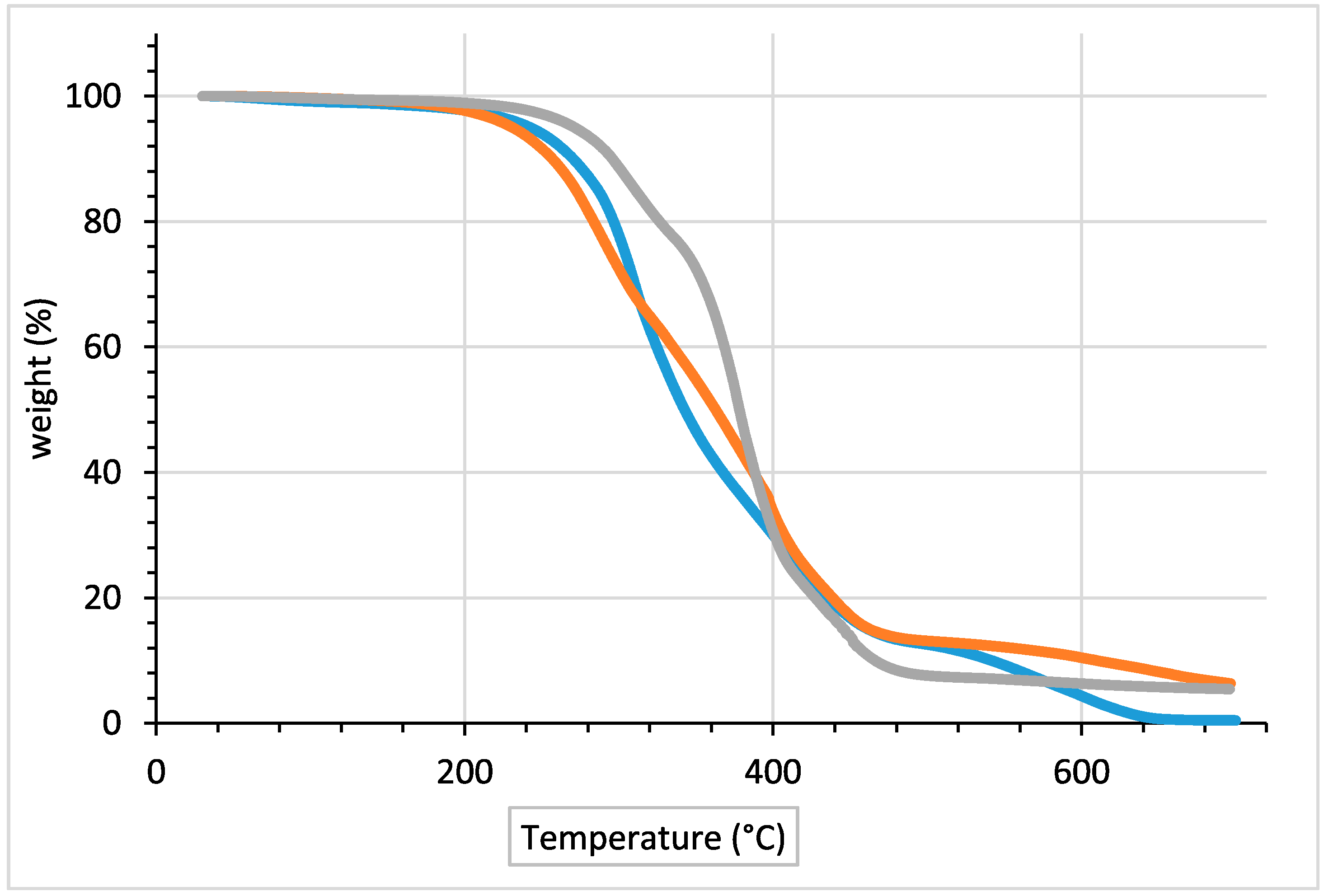
TGA curves of polyurethane samples obtained using 1 mol % and 2 mol % of Mo catalyst and literature procedure, analyzed by the first derivative (DTG), showed a multistep decomposition pattern in agreements with literature (
Figure 5) [
40,
41,
42,
43]. At temperature lower than 200 °C the weight loss was due to small molecule products (not reacted isocyanates, water, ethers, etc.).
The urethane bond groups break up into isocyanates and polyols from 200 °C. At the same time, the polyols segments decompose to same kinds of aliphatic alcohols and the products become more complex as the evolved products interact with each other. At temperature above 350 °C, primary and secondary amines, vinyl ethers, CO
2, HCN, and nitriles are the decomposition products [
42,
43].
The values of degradation temperatures for 5% weight loss (T
d5%), for 70% weight loss (T
d70%), for 90% weight loss (T
d90%), the maximum degradation temperature (Tmax) and the char yield at 700 °C are shown in
Table 2.
These results indicate that the polyurethane sample prepared using the literature procedure is initially the most stable. In fact, the 5% degradation weight loss is obtained at 272.1 °C, thirty and forty degrees above the weight loss temperatures observed for Bio-PU samples deriving from 1 mol % and 2 mol % catalyst. Moreover, temperatures at which the degradation process shows the maximum rate follow the same order: 291.7 °C for 2 mol % Bio-PU sample, 312.4 °C for 1 mol % Bio-PU sample and 378.3 °C for literature samples.
At about the 70% weight loss the situation changes. The literature polyurethane sample becomes less stable than polyurethane samples prepared using the one-pot synthesis herein reported. In particular, the Bio-PU with 2 mol % MoCl
2O
2 catalyst has the highest temperature at which the 90% degradations weight loss is obtained. However, the shapes of the weight loss curves of all polyurethanes are almost identical, and overall differences in thermal stability appear to be small [
44,
45].


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}