Recent Advances in Organocatalyzed Domino C–C Bond-Forming Reactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mannich
3. Henry Reactions
4. Aldol Reactions
5. Other Reactions
5.1. Knoevenegal/Diels-Alder Reactions
5.2. Wittig Reactions
Acknowledgments
Conflicts of Interest
References
- Scheffler, U.; Mahrwald, R. Recent Advances in Organocatalytic Methods for Asymmetric C–C Bond Formation. Chem. Eur. J. 2013, 19, 14346–14396. [Google Scholar] [CrossRef] [PubMed]
- Rue**, M.; Atodiresei, I. 6.13 C–C Bond Formation: Cascade or Domino Reaction. In Comprehensive Chirality; Elsevier: Amsterdam, The Netherlands, 2012; pp. 345–373. ISBN 978-0-08-095168-3. [Google Scholar]
- Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances. Molecules 2017, 22, 1636. [Google Scholar] [CrossRef] [PubMed]
- Arend, M.; Westermann, B.; Risch, N. Modern Variants of the Mannich Reaction. Angew. Chem. Int. Ed. 1998, 37, 1044–1070. [Google Scholar] [CrossRef]
- Córdova, A. The Direct Catalytic Asymmetric Mannich Reaction. Acc. Chem. Res. 2004, 37, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Verkade, J.M.M.; van Hemert, L.J.C.; Quaedflieg, P.J.L.M.; Rutjes, F.P.J.T. Organocatalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Song, B.; Bhadury, P.S.; Li, L.; Wang, Z.; Zhang, X.; Hu, D.; Chen, Z.; Zhang, Y.; Bai, S.; et al. Chiral Cinchona Alkaloid-Derived Thiourea Catalyst for Enantioselective Synthesis of Novel β-amino Esters by Mannich Reaction. Chirality 2012, 24, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, G.S. Recent Applications of Cinchona Alkaloids And Their Derivatives As Catalysts In Metal-Free Asymmetric Synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Poulsen, T.B.; Alemparte, C.; Saaby, S.; Bella, M.; Jørgensen, K.A. Direct Organocatalytic and Highly Enantio- and Diastereoselective Mannich Reactions of α-Substituted α-Cyanoacetates. Angew. Chem. Int. Ed. 2005, 44, 2896–2899. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Li, J.-L.; ** Novel Organocatalyzed Aldol Reactions for the Enantioselective Synthesis of Biologically Active Molecules. Synthesis 2011, 2011, 1815–1830. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Boomhoff, M. Aldol Reactions in Domino Processes. In Domino Reactions; Tietze, L.F., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 267–294. ISBN 978-3-527-67130-4. [Google Scholar]
- Andrushko, V.; Andrushko, N. (Eds.) Stereoselective Synthesis of Drugs and Natural Products; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-118-03217-6. [Google Scholar]
- Meazza, M.; Potter, M.; Pitak, M.B.; Coles, S.J.; Mazzanti, A.; Rios, R. Highly Enantioselective Synthesis of Alkylpyridine Derivatives through a Michael/Michael/Aldol Cascade Reaction. Eur. J. Org. Chem. 2017, 2017, 719–725. [Google Scholar] [CrossRef]
- Kumar, M.; Chauhan, P.; Valkonen, A.; Rissanen, K.; Enders, D. Asymmetric Synthesis of Functionalized Tricyclic Chromanes via an Organocatalytic Triple Domino Reaction. Org. Lett. 2017, 19, 3025–3028. [Google Scholar] [CrossRef] [PubMed]
- Simon-Levert, A.; Arrault, A.; Bontemps-Subielos, N.; Canal, C.; Banaigs, B. Meroterpenes from the Ascidian Aplidium aff. densum. J. Nat. Prod. 2005, 68, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Garrido, L.; Zubía, E.; Ortega, M.J.; Salvá, J. New Meroterpenoids from the Ascidian Aplidium conicum. J. Nat. Prod. 2002, 65, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Am Ende, C.W.; Zhou, Z.; Parker, K.A. Total Synthesis of (±)-Bisabosqual A. J. Am. Chem. Soc. 2013, 135, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Skrabek, R.Q.; Galimova, L.; Ethans, K.; Perry, D. Nabilone for the Treatment of Pain in Fibromyalgia. J. Pain 2008, 9, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Li, X.-C.; Dunbar, D.C.; ElSohly, M.A.; Khan, I.A. Antimalarial (+)-trans-Hexahydrodibenzopyran Derivatives from Machaerium multiflorum. J. Nat. Prod. 2001, 64, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Li, X.-C.; Jacob, M.R.; Tekwani, B.L.; Dunbar, D.C.; Ferreira, D. Antimicrobial and Antiparasitic (+)-trans-Hexahydrodibenzopyrans and Analogues from Machaerium multiflorum. J. Nat. Prod. 2003, 66, 804–809. [Google Scholar] [CrossRef] [PubMed]
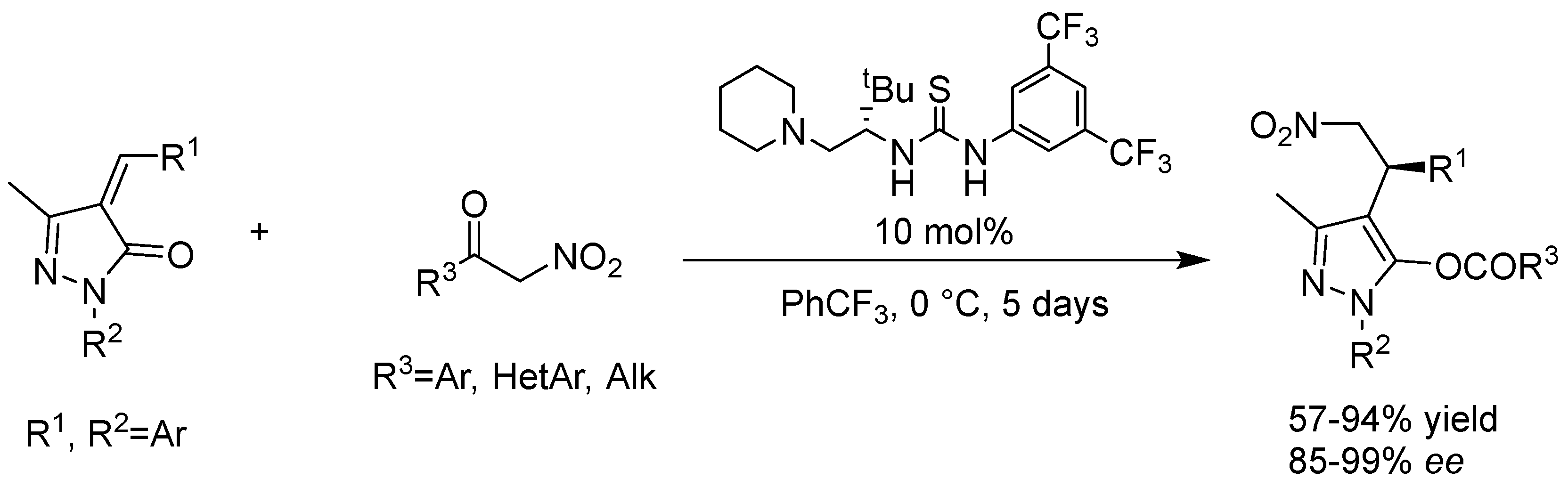
- Maity, R.; Gharui, C.; Sil, A.K.; Pan, S.C. Organocatalytic Asymmetric Michael/Hemiketalization/Retro-aldol Reaction of α-Nitroketones with Unsaturated Pyrazolones: Synthesis of 3-Acyloxy Pyrazoles. Org. Lett. 2017, 19, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Dreger, A. Recent Advances in the Chemistry of Pyrazoles. Properties, Biological Activities, and Syntheses. Curr. Org. Chem. 2011, 15, 1423–1463. [Google Scholar] [CrossRef]
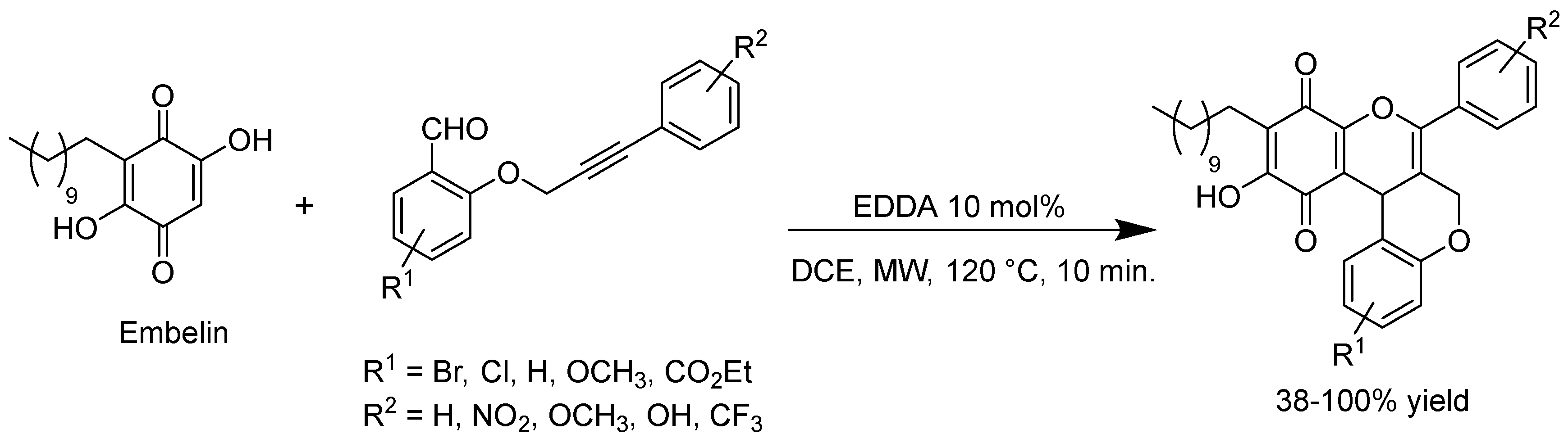
- Martín-Acosta, P.; Feresin, G.; Tapia, A.; Estévez-Braun, A. Microwave-Assisted Organocatalytic Intramolecular Knoevenagel/Hetero Diels–Alder Reaction with O-(Arylpropynyloxy)-Salicylaldehydes: Synthesis of Polycyclic Embelin Derivatives. J. Org. Chem. 2016, 81, 9738–9756. [Google Scholar] [CrossRef] [PubMed]
- Peña, R.; Martín, P.; Feresin, G.E.; Tapia, A.; Machín, F.; Estévez-Braun, A. Domino Synthesis of Embelin Derivatives with Antibacterial Activity. J. Nat. Prod. 2016, 79, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, J.; Wang, Y.; Qiao, L.; Zhou, Y. Embelin and Its Role in Chronic Diseases. In Anti-inflammatory Nutraceuticals and Chronic Diseases; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2016; pp. 397–418. ISBN 978-3-319-41332-7. [Google Scholar]
- Enders, D.; Grossmann, A.; Gieraths, B.; Düzdemir, M.; Merkens, C. Organocatalytic One-Pot Asymmetric Synthesis of 4H,5H-Pyrano[2,3-c]pyrazoles. Org. Lett. 2012, 14, 4254–4257. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.-C.; Jan, R.-H.; Tsai, C.-W.; Nimje, R.Y.; Liao, J.-H.; Lee, G.-H. Organocatalytic Enantioselective Cascade Michael−Michael−Wittig Reactions of Phosphorus Ylides: One-Pot Synthesis of the all-cis Trisubstituted Cyclohexenecarboxylates via the [1 + 2 + 3] Annulation. Org. Lett. 2009, 11, 5246–5249. [Google Scholar] [CrossRef] [PubMed]
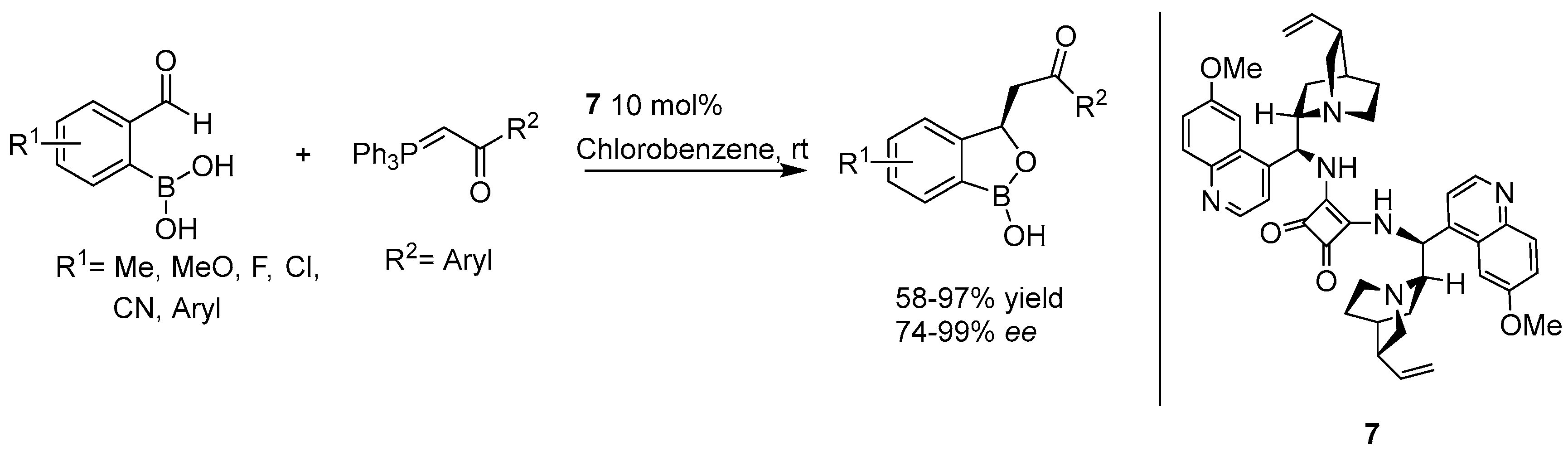
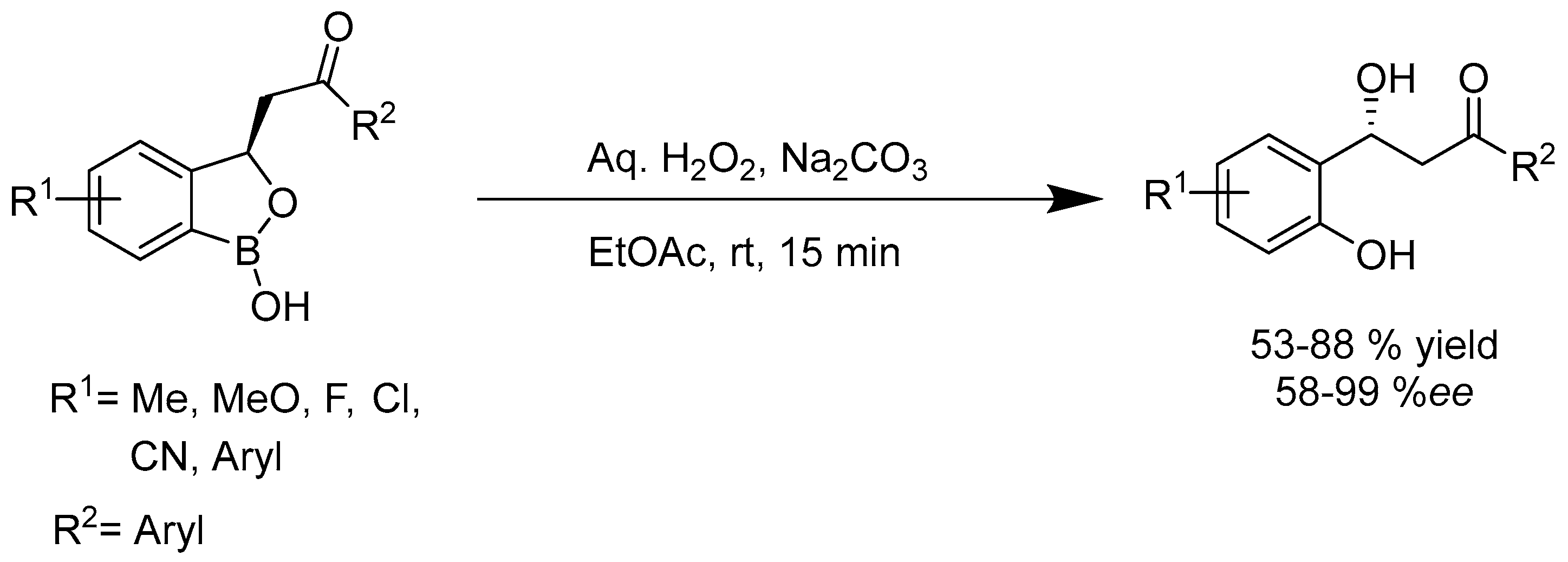
- Hazra, G.; Maity, S.; Bhowmick, S.; Ghorai, P. Organocatalytic, enantioselective synthesis of benzoxaboroles via Wittig/oxa-Michael reaction Cascade of α-formyl boronic acids. Chem. Sci. 2017, 8, 3026–3030. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent Developments in the Chemistry and Biological Applications of Benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef] [PubMed]














© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, C.S.; Davis, L.O. Recent Advances in Organocatalyzed Domino C–C Bond-Forming Reactions. Molecules 2018, 23, 33. https://doi.org/10.3390/molecules23010033
Evans CS, Davis LO. Recent Advances in Organocatalyzed Domino C–C Bond-Forming Reactions. Molecules. 2018; 23(1):33. https://doi.org/10.3390/molecules23010033
Chicago/Turabian StyleEvans, Cleo S., and Lindsey O. Davis. 2018. "Recent Advances in Organocatalyzed Domino C–C Bond-Forming Reactions" Molecules 23, no. 1: 33. https://doi.org/10.3390/molecules23010033