
Conformational and Tautomeric Control by Supramolecular Approach in Ureido-N-iso-propyl,N’-4-(3-pyridin-2-one)pyrimidine

Abstract
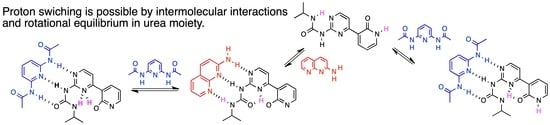
:
1. Introduction
2. Results and Discussion
2.1. NMR Measurements
2.2. 1H VT (Variable Temperature) NMR
2.3. Guest Replacement
2.4. DFT Calculations
3. Materials and Methods
3.1. Synthesis
3.2. Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beijer, F.H.; Sijbesma, R.P.; Kooijman, H.; Spek, A.L.; Meijer, E.W. Strong Dimerization of Ureidopyrimidones via Quadruple Hydrogen Bonding. J. Am. Chem. Soc. 1998, 120, 6761–6769. [Google Scholar] [CrossRef]
- Scherman, O.A.; Ligthart, G.B.W.L.; Ohkawa, H.; Sijbesma, R.P.; Meijer, E.W. Olefin metathesis and quadruple hydrogen bonding: A powerful combination in multistep supramolecular synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 11850–11855. [Google Scholar] [CrossRef] [Green Version]
- Kolehmainen, E.; Ośmiałowski, B.; Nissinen, M.; Kauppinen, R.; Gawinecki, R. Substituent and temperature controlled tautomerism of 2-phenacylpyridine: The hydrogen bond as a configurational lock of (Z)-2-(2-hydroxy-2-phenylvinyl)pyridine. J. Chem. Soc. Perkin Trans. 2 2000, 11, 2185–2191. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Mroczyńska, K.; Kolehmainen, E.; Kowalska, M.; Valkonen, A.; Pietrzak, M.; Rissanen, K. Association of N-(pyridin-2-yl),N′-substituted ureas with 2-amino-1,8-naphthyridines and benzoates: NMR and quantum chemical studies of the substituent effect on complexation. J. Org. Chem. 2013, 78, 7582–7593. [Google Scholar] [CrossRef]
- Aprahamian, I. Molecular machines: Molecules bearing robotic arms. Nat. Chem. 2016, 8, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.L.; Zhong, J.Q.; Lin, J.D.; Hu, W.P.; Wu, K.; Xu, G.Q.; Wee, A.T.S.; Chen, W. Towards single molecule switches. Chem. Soc. Rev. 2015, 44, 2998–3022. [Google Scholar] [CrossRef] [Green Version]
- Draper, E.R.; Eden, E.G.B.; McDonald, T.O.; Adams, D.J. Spatially resolved multicomponent gels. Nat. Chem. 2015, 7, 848. [Google Scholar] [CrossRef]
- Nevo, R.; Stroh, C.; Kienberger, F.; Kaftan, D.; Brumfeld, V.; Elbaum, M.; Reich, Z.; Hinterdorfer, P. A molecular switch between alternative conformational states in the complex of Ran and importin β1. Nat. Struct. Mol. Biol. 2003, 10, 553. [Google Scholar] [CrossRef]
- Mohn, F.; Repp, J.; Gross, L.; Meyer, G.; Dyer, M.S.; Persson, M. Reversible Bond Formation in a Gold-Atom--Organic-Molecule Complex as a Molecular Switch. Phys. Rev. Lett. 2010, 105, 266102. [Google Scholar] [CrossRef]
- Kokan, Z.; Chmielewski, M.J. A Photoswitchable Heteroditopic Ion-Pair Receptor. J. Am. Chem. Soc. 2018, 140, 16010–16014. [Google Scholar] [CrossRef] [PubMed]
- Epa, K.; Aakeröy, C.B.; Desper, J.; Rayat, S.; Chandra, K.L.; Cruz-Cabeza, A.J. Controlling molecular tautomerism through supramolecular selectivity. Chem. Commun. 2013, 49, 7929–7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Xu, J.; Sun, Z.; Fu, B.; Qin, C.; Zeng, L.; Hu, X. A twisted intramolecular charge transfer probe for rapid and specific detection of trace biological SO2 derivatives and bio-imaging applications. Chem. Commun. 2015, 51, 1154–1156. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Nakamura, T.; Yoshioka, M.; Shimomura, Y. A molecular switch based on acid and base promoted, cation governed binding in a crown ether threaded rotaxane. Tetrahedron Lett. 2006, 47, 5901–5904. [Google Scholar] [CrossRef]
- Grunder, S.; McGrier, P.L.; Whalley, A.C.; Boyle, M.M.; Stern, C.; Stoddart, J.F. A Water-Soluble pH-Triggered Molecular Switch. J. Am. Chem. Soc. 2013, 135, 17691–17694. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wu, Y.; Wang, Y.; Liu, Z.; Cheng, C.; Hartlieb, K.J.; Wasielewski, M.R.; Stoddart, J.F. An Electrochromic Tristable Molecular Switch. J. Am. Chem. Soc. 2015, 137, 13484–13487. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Kagawa, F.; Hatahara, K.; Kobayashi, K.; Kumai, R.; Murakami, Y.; Tokura, Y. Above-room-temperature ferroelectricity and antiferroelectricity in benzimidazoles. Nat. Commun. 2012, 3, 1308. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, S.; Kumai, R.; Tokura, Y. Hydrogen-Bonding Molecular Chains for High-Temperature Ferroelectricity. Adv. Mater. 2011, 23, 2098–2103. [Google Scholar] [CrossRef]
- Antonov, L.; Deneva, V.; Simeonov, S.; Kurteva, V.; Nedeltcheva, D.; Wirz, J. Exploiting Tautomerism for Switching and Signaling. Angew. Chem. 2009, 121, 8015–8018. [Google Scholar] [CrossRef]
- Kolehmainen, E.; Ośmialowski, B.; Krygowski, T.M.; Kauppinen, R.; Nissinen, M.; Gawinecki, R. Substituent and temperature controlled tautomerism: Multinuclear magnetic resonance, X-ray, and theoretical studies on 2-phenacylquinolines. J. Chem. Soc. Perkin Trans. 2 2000, 6, 1259–1266. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the.beta.-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Resonance-Assisted Hydrogen Bonds: A Critical Examination. Structure and Stability of the Enols of β-Diketones and β-Enaminones. J. Phys. Chem. A 2007, 111, 3585–3591. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wan, W.; Yong, X.; Lu, X.; Liu, R.; Qu, J. Urea-based polyacetylenes as an optical sensor for fluoride ions. Chinese J. Polym. Sci. 2013, 31, 620–629. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Wittkopp, A. H-Bonding Additives Act Like Lewis Acid Catalysts. Org. Lett. 2002, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Howard, J.A.K.; Steed, J.W. Anion-switchable supramolecular gels for controlling pharmaceutical crystal growth. Nat. Chem. 2010, 2, 1037. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yip, W.H.; Mak, T.C.W. Hydrogen-bonded urea-anion host lattices. Part 2. Crystal structures of inclusion compounds of urea with tetraalkylammonium bicarbonates. J. Inclus. Phenom. Mol. 1995, 23, 233–244. [Google Scholar] [CrossRef]
- Kushner, A.M.; Vossler, J.D.; Williams, G.A.; Guan, Z. A Biomimetic Modular Polymer with Tough and Adaptive Properties. J. Am. Chem. Soc. 2009, 131, 8766–8768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Greef, T.F.A.; Smulders, M.M.J.; Wolffs, M.; Schenning, A.P.H.J.; Sijbesma, R.P.; Meijer, E.W. Supramolecular Polymerization. Chem. Rev. 2009, 109, 5687–5754. [Google Scholar] [CrossRef]
- Mroczyńska, K.; Kaczorowska, M.; Kolehmainen, E.; Grubecki, I.; Pietrzak, M.; Ośmiałowski, B. Conformational equilibrium in supramolecular chemistry: Dibutyltriuret case. Beilstein J. Org. Chem. 2015, 11, 2105–2116. [Google Scholar] [CrossRef] [Green Version]
- Corbin, P.S.; Zimmerman, S.C.; Thiessen, P.A.; Hawryluk, N.A.; Murray, T.J. Complexation-induced unfolding of heterocyclic ureas. Simple foldamers equilibrate with multiply hydrogen-bonded sheetlike structures. J. Am. Chem. Soc. 2001, 123, 10475–10488. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Kolehmainen, E. Comment on “non-symmetric substituted ureas locked in an (E,Z) conformation: An unusual anion binding via supramolecular assembly” by M. Olivari, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, R. Montis and M. A. Scorciapino, New J. Chem. 2013, 37, 663. New J. Chem. 2014, 38, 2701–2703. [Google Scholar]
- Pellizzaro, M.L.; McGhee, A.M.; Renton, L.C.; Nix, M.G.; Fisher, J.; Turnbull, W.B.; Wilson, A.J. Conformer-independent ureidoimidazole motifs-tools to probe conformational and tautomeric effects on the molecular recognition of triply hydrogen-bonded heterodimers. Chem. Eur. J. 2011, 17, 14508–14517. [Google Scholar] [CrossRef] [PubMed]
- Corbin, P.S.; Zimmerman, S.C. Self-association without regard to prototropy. A heterocycle that forms extremely stable quadruply hydrogen-bonded dimers. J. Am. Chem. Soc. 1998, 120, 9710–9711. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kolehmainen, E.; Loghmani-Khouzani, H.; Ośmiałowski, B.; Lovász, T.; Rosa, P. Effect of π-electron delocalization on tautomeric equilibria - Benzoannulated 2-phenacylpyridines. Eur. J. Org. Chem. 2006, 2006, 2817–2824. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kolehmainen, E.; Zakrzewski, A.; Laihia, K.; Ośmiałowski, B.; Kauppinen, R. Predominance of inductive over resonance substituent effect on 33S NMR chemical shifts of 4-substituted phenyl-4′-methylphenacyl sulfones. Magn. Reson. Chem. 1999, 37, 437–440. [Google Scholar] [CrossRef]
- Ośmiałowski, B. Proton transfer reaction and intermolecular interactions in associates of 2,5-dihydroxy-1,8-naphthyridine. J. Mol. Model. 2012, 18, 1633–1644. [Google Scholar] [CrossRef]
- Dobosz, R.; Ośmiałowski, B.; Gawinecki, R. DFT studies on tautomeric preferences. Part 3: Proton transfer in 2-(8-acylquinolin-2-yl)-1,3-diones. Struct. Chem. 2010, 21, 1037–1041. [Google Scholar] [CrossRef] [Green Version]
- Dobosz, R.; Skotnicka, A.; Rozwadowski, Z.; Dziembowska, T.; Gawinecki, R. Stability of N-(ortho-hydroxynaphthylmethylene)methylamines and their tautomers. J. Mol. Struct. 2010, 979, 194–199. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Maine, F.W.; Golding, S. The Tautomerism of heteroaromatic compounds with five-membered rings—IX: N-unsubstituted pyrazolin-3(5)-ones. Tetrahedron 1965, 21, 1693–1699. [Google Scholar] [CrossRef]
- Frank, J.; Katritzky, A.R. Tautomeric pyridines. Part XV. Pyridone-hydroxypyridine equilibria in solvents of differing polarity. J. Chem. Soc. Perkin Trans. 2 1976, 12, 1428–1431. [Google Scholar] [CrossRef]
- Vallejo Narváez, W.E.; Jiménez, E.I.; Romero-Montalvo, E.; Sauza-de la Vega, A.; Quiroz-García, B.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Acidity and basicity interplay in amide and imide self-association. Chem. Sci. 2018, 9, 4402–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Brynn Hibbert, D.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ośmiałowski, B.; Kolehmainen, E.; Ikonen, S.; Valkonen, A.; Kwiatkowski, A.; Grela, I.; Haapaniemi, E. 2-acylamino- and 2,4-bis(acylamino)pyrimidines as supramolecular synthons analyzed by multiple noncovalent interactions. DFT, X-ray diffraction, and NMR spectral studies. J. Org. Chem. 2012, 77, 9609–9619. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.H.; Facelli, J.C.; de Kowalewski, D.G. A proton NMR analysis of the OCH3 group conformation in 2-methoxypyridines. Org. Magn. Res. 1982, 20, 40–41. [Google Scholar] [CrossRef]
- Benesi, H.; Hildebrand, J. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, J.; Tang, B.Z. Kinetic trap** – a strategy for directing the self-assembly of unique functional nanostructures. Chem. Commun. 2016, 52, 11870–11884. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Espinosa, E.; Souhassou, M.; Lachekar, H.; Lecomte, C. Topological analysis of the electron density in hydrogen bonds. Acta Cryst. B 1999, 55, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Storch, G.; Spallek, M.J.; Rominger, F.; Trapp, O. Tautomerization-Mediated Molecular Switching Between Six- and Seven-Membered Rings Stabilized by Hydrogen Bonding. Chem. Eur. J. 2015, 21, 8939–8945. [Google Scholar] [CrossRef]
- Matsumoto, J.; Ishizu, M.; Kawano, R.; Hesaka, D.; Shiragami, T.; Hayashi, Y.; Yamashita, T.; Yasuda, M. Generation of quinone methide from aminomethyl(hydroxy)arenes precursors in aqueous solution. Tetrahedron 2005, 61, 5735–5740. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02. Inc. Wallingford CT 2009, 200, 28. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp./Probe b | H5 | H6 | H9 | H13 | H15 |
|---|---|---|---|---|---|
| 1 + A | 608 (7 c) | 448 (4) | 767 (10) | 699 (11) | |
| 1 + B | 1656 (15) | 1253 (10) | 932(14) | ||
| 1Me + B | 162 (4) | 110 (4) | 125 (4) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiatkowski, A.; Kolehmainen, E.; Ośmiałowski, B. Conformational and Tautomeric Control by Supramolecular Approach in Ureido-N-iso-propyl,N’-4-(3-pyridin-2-one)pyrimidine. Molecules 2019, 24, 2491. https://doi.org/10.3390/molecules24132491
Kwiatkowski A, Kolehmainen E, Ośmiałowski B. Conformational and Tautomeric Control by Supramolecular Approach in Ureido-N-iso-propyl,N’-4-(3-pyridin-2-one)pyrimidine. Molecules. 2019; 24(13):2491. https://doi.org/10.3390/molecules24132491
Chicago/Turabian StyleKwiatkowski, Adam, Erkki Kolehmainen, and Borys Ośmiałowski. 2019. "Conformational and Tautomeric Control by Supramolecular Approach in Ureido-N-iso-propyl,N’-4-(3-pyridin-2-one)pyrimidine" Molecules 24, no. 13: 2491. https://doi.org/10.3390/molecules24132491