2.1. BODIPYs
Since their first report in 1968 by Treibs and Kreuzer, 4,4-difluoro-4-bora-diazaindacenes (BODIPYs) have been thoroughly studied and are now considered to be one of the most important families of organic fluorophores [
50]. Locking of the cyanine monomethine backbone by the boron atom confers to BODIPYs excellent (photo)stability, high extinction coefficients (ε), high quantum yields (Φ
F) in solution, sharp absorption bands, and sharp emission bands, positioning them as a dye of choice for many applications [
51,
52]. Moreover, possible NIRF emission and global neutrality of these dyes are of particular interest for imaging purposes. One drawback of BODIPY dyes is their low Stokes shifts (usually below 1000 cm
−1), reducing signal-to-background ratio due to reabsorption and use of narrow filters. Nowadays, biomedical applications of BODIPY dyes are mainly focused on cellular imaging and sensing, however, examples of NIRF OPI and photoacoustic imaging (PAI) have been described [
53,
54]. Given the importance of BODIPY dyes as fluorescent probes, several groups have been involved in the development of dual-mode imaging agents including OPI-PET, OPI-SPECT, or OPI-MRI [
53,
55,
56]. Recently, Ali et al. published an overview of radiolabeled BODIPYs in the literature including
125I,
111In, and
89Zr adducts [
57]. The majority of
18F-BODIPYs are labeled on the boron center (
Scheme 1), however, the first example reported by Perrin et al. describes radiolabeling via the use of a boronic ester [
58]. This work is one of the first of a series on boronic ester-mediated
18F radiolabeling. This method is more often used for the radiosynthesis of xanthene- and cyanine-based tracers and is further detailed in the related sections (vide infra). Other methods rely on the presence of the central boron atom and can be classified into three types, one based on two or three steps labeling through an activated intermediate and the others based either on hydroxy substitution or on direct isotopic exchange (IEX), both acid-catalyzed.
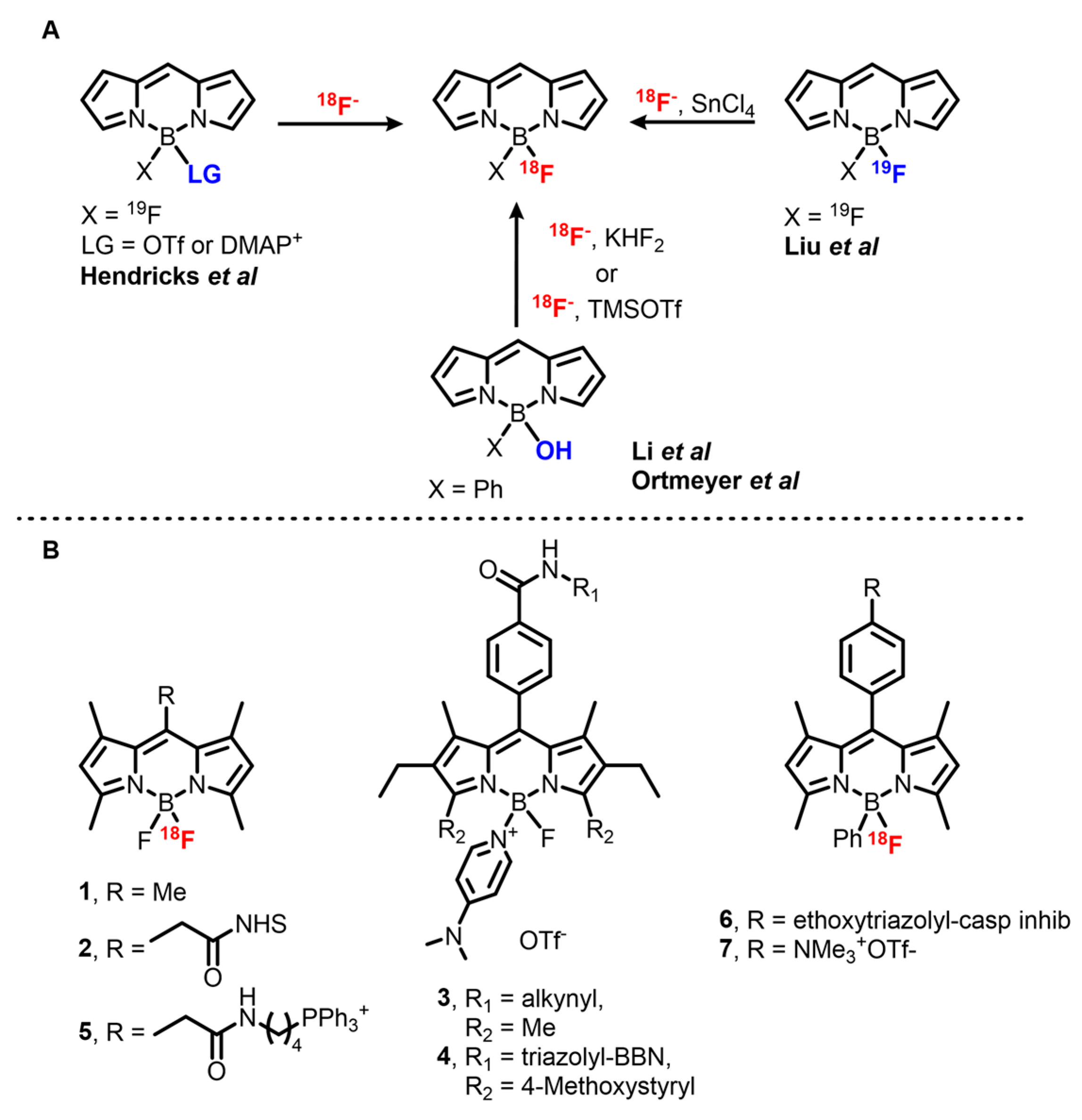
Development on the first method started in 2008 when Hudnall and Gabbaï described a BODIPY-4-dimethylaminopyridine (DMAP) cation for turn-on sensing of fluoride by displacement of DMAP from the boron atom [
62]. This procedure was adapted for preparative purposes in 2012 by Hendricks et al. and afforded [
18F]BODIPY
1 with a 68 ± 23% d.c. RCY [
59]. However, the high temperature required for the fluoride–DMAP exchange is incompatible with sensitive moieties, especially bioconjugation handles such as
N-hydroxysuccinimide (NHS) esters. Moreover, upon extended heating or in the presence of acid, product decomposition and exchange of
18F with released
19F can occur, decreasing the yield and molar activity of the product. This issue was addressed by a one-pot radiolabeling procedure through activation of [
19F]BODIPY
1 with TMSOTf and direct exchange with
18F in presence of TfOH. The two reactions occur quickly at 20 °C and the intermediate can be stabilized in solution by buffering the media with a mild, non-nucleophilic base such as DIPEA or 2,6-lutidine. This modified method afforded [
18F]
1 within 2 min in 67% RCY, with A
m = 0.96 Ci/μmol. Following a slightly modified method, NHS ester [
18F]
2 was obtained and subsequently coupled with the monoclonal antibody trastuzumab with a 19.9% d.c. RCY. Soon after, Yuan et al. used this method to synthesize OPI/PET probes for mitochondria imaging by coupling [
18F]
1 with an amine bearing triphenylphosphonium derivative to afford [
18F]
5 with A
m = 0.37 mCi/μmol [
63].
In 2014, Weissleder’s group investigated a one-step version of this reaction by sequentially adding Tf
2O,
tBuOH, and a BF
2-BODIPY precursor to the
18F/
n-tetrabutyl ammonium bromide (TBAB) mix [
64]. A wide range of commercially available BODIPY-NHS esters and PARPi-FL, a PARP1 inhibitor currently studied for detection of epithelial cancers, were labeled with this method [
65]. However, despite the efforts invested in the study of this reaction, the presence of unreacted starting material in the reaction media limited A
m values to a range of 5–80 mCi/μmol for radiolabeling of [
18F]
1 and therefore dampened the use of this procedure for radiopharmaceutical production.
In 2014, Brizet et al. used the BODIPY-DMAP stability to synthesize a series of PET imaging precursors [
66]. They showed that the DMAP adduct withstands Huisgen cycloaddition conditions, isothiocyanate synthesis, and their subsequent coupling with amines. Green emitting BODIPY-DMAP
3 bearing a terminal alkyne was successfully coupled with peptides such as bombesin, and the DMAP part was successfully displaced using KF, suggesting that BODIPYs could be used as prosthetic groups for late-stage peptide radiolabeling. However, a major side product of this reaction is the BF(OH) complex stemming from hydrolysis of the DMAP group.
Simultaneously with the development of this first method, Gabbaï et al. described an acid-mediated substitution of a hydroxy group attached to the central boron atom of the BODIPY core with fluorine by treatment of a B(Ph)(OH)BODIPY with KHF
2 in THF [
67]. This method was then modified to allow
18F labeling in aqueous conditions. [
18F]BODIPY
7 was obtained in 22 ± 3% d.c. RCY and a A
m > 1.4 Ci/μmol without preliminary
18F drying step by reaction of the precursor with KHF
2 in a MeOH/[
18O]H
2O mix at 70 °C for 10 min [
42]. Ortmeyer et al. used a slightly simplified method to label an isatin-based caspase inhibitor [
18F]
6 in 11% ± 6.1% d.c. RCY in 91 ± 6 min with a high molar activity (A
m > 4.48 Ci/μmol) [
60].
Finally, Lewis acid-promoted IEX on BODIPY dyes, developed in 2013 by Gabbaï et al., is the most used reaction for
18F labeling of this family of dyes [
61], possibly due to ready implementation and the ability to afford an extended range of tracers in high yields. Among the set of Lewis acids used, SnCl
4 in excess (8 to 14 molar excess in this case) was shown to be the most effective, affording [
18F]
7 with a nearly quantitative yield (95% d.c. RCY) in ACN at room temperature and in only 10 min. This method is compatible with NHS esters, making it attractive for the synthesis of imaging probes. Other bioconjugation groups are tolerated as has been demonstrated for terminal alkynes and azide moieties. In addition, BODIPY conjugates with small peptides such as RGD, an α
Vβ
3 integrin receptor ligand used for cancer imaging, can be obtained in very good RCY (82%). In addition, BODIPYs bearing various substituents such as alkyls, aryls, and styryls afforded OPI/PET probes with emission bands between 500 and 650 nm. The downside of this method resides in the low molar activity of the products, which are usually in the mCi/μmol range. This is likely due to the presence of remaining starting material sharing the exact same structure as the product in the reaction mixture and thus being impossible to remove. However, it appears that the use of [
18F]BODIPYs afforded via this method has not been hindered. Indeed, this method was applied to the synthesis of [
18F]BODIPY radiotracers bearing various functional groups, peptides (RGD, RGD dimers, bombesin, MT-MMP1 substrates), different small molecule-based inhibitors (PARP1, PACMA31), and lipids either at late stage or before conjugation [
55,
68,
69,
70,
71,
72,
73].
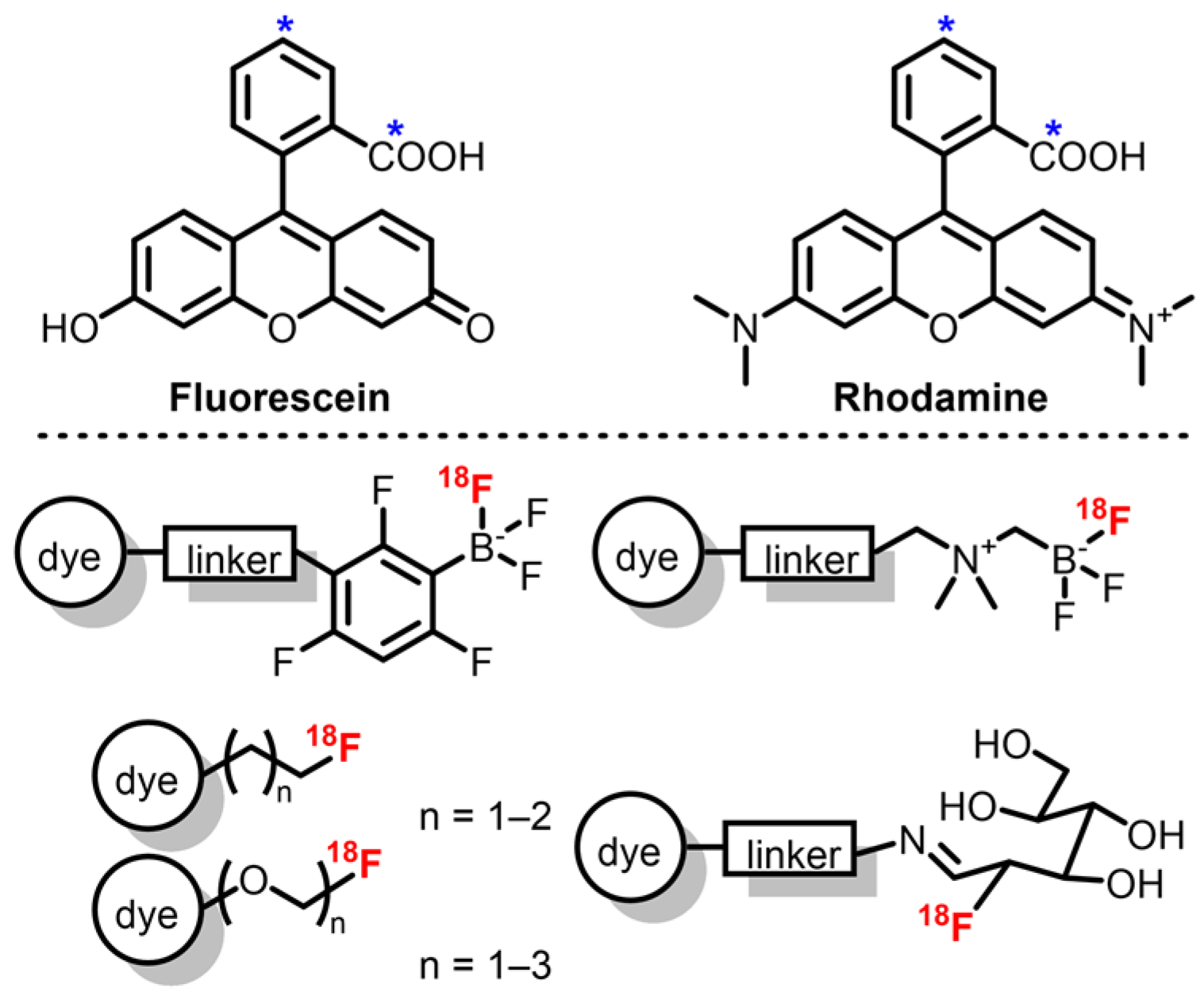
2.2. Fluoresceins and Rhodamines
Similar to BODIPYs and cyanines, xanthene derivatives including fluorescein and rhodamine are remarkable fluorophores with high Φ
F and ε, and good photostability [
74,
75]. Compared to the former families, their major difference resides in the presence of an equilibrium (influenced by solvent and pH) between the closed spiro-lactone form, which is non-emissive, and the open emissive form, allowing development of environmentally sensitive probes. Regarding OPI, alkylated rhodamine derivatives are usually preferred as their emission band is red-shifted compared to fluoresceins (λ
em around 570 nm and 510 nm, respectively) and they present an improved photostability [
74]. Despite extensive work on fluorescein and rhodamine synthesis and functionalization, emission maxima above 700 nm are rarely described, which can be a disadvantage when compared to previous dyes. However, these dyes are well suited for dual mode PET/OPI probes as their phenyl moiety represent a good platform for introduction of
18F by use of a prosthetic group. In the case of xanthene derivatives, these groups include mainly [
18F]fluoroethyl tosylate derivatives and boron trifluoride derivatives (
Scheme 2).
The first examples of
18F-labeled rhodamine were described by Packard et al., who synthesized [
18F]fluoroethyl rhodamine-B via nucleophilic substitution on [
18F]fluoroethyl tosylate by the rhodamine’s carboxylic acid group [
76,
77]. This two-step procedure afforded the probe with a 35% d.c. RCY in 90 min with A
m = 35 mCi/μmol. Purification of the intermediate [
18F]fluoroethyl tosylate allowed A
m to reach 67 mCi/μmol. Soon after, the authors also studied the effect of the prosthetic group on the pharmacokinetics of rhodamine-B by replacing the fluoroethyl group by fluoropropyl, fluoro di-, and fluoro tri-ethyleneglycol groups [
78]. Lower A
m was observed due to side reactions with remaining ditosylate precursors. This methodology was also applied to rhodamine-4Me, -6G, and -101 in similar RCY and A
m [
79]. Finally, replacement of the one-pot two-steps procedure by the synthesis of the tosylated diethylene glycol ester of rhodamine-6G readily labeled with
18F afforded the corresponding tracer with A
m up to 7 Ci/μmol [
80]. RCYs were unfortunately decreased (6 ± 3% d.c.) due to ineffective elution of
18F ions from the QMA cartridge and partial retention of the lipophilic tracer on the synthesizer’s tubing. However, this method represents an easy way to afford
18F-rhodamines with high molar activities.
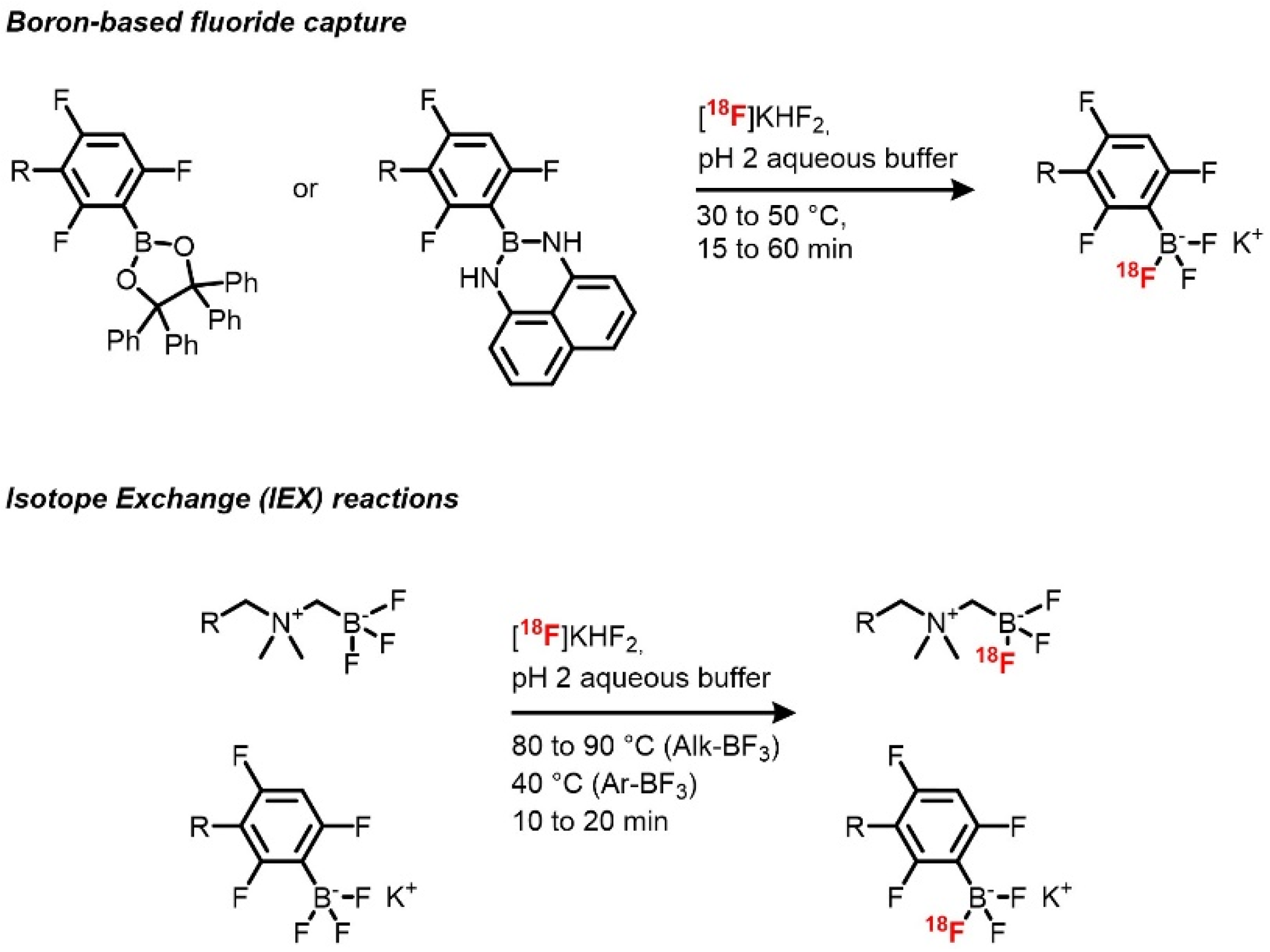
18F boron trifluoride xanthene derivatives can be achieved by fluoride capture by a boron-containing reactive group and by IEX on boron trifluoride species (
Scheme 3) [
81,
82]. The use of inorganic fluoride captors has been applied to radiochemistry in order to allow late-stage labeling of biomolecules with high molar activities, and boron-based prosthetic groups have been used in the design of several prosthetic groups [
83,
84]. Radiosynthesis using the IEX reactions has been previously demonstrated on organic molecules, then followed by silicon- and boron-based prosthetic groups. Boron trifluoride derivatives as radiolabeling precursors of fluorescent compounds were first described by the team of Perrin. These groups proved to be useful for late-stage radiolabeling thanks to the high rates of exchange and the slightly acidic reaction conditions, allowing their use on a wide range of compounds. In the case of xanthene dyes, [
18F]sulforhodamine B was quickly (10–15 min of reaction time) afforded with d.c. RCYs between 45 and 75%, high RCPs, and very high A
m (up to 14.3 Ci/μmol), allowed also by the use of a low amount of precursor [
81]. Other advantages of this method are the absence of
18F drying step and prolonged heating (40 °C), which respectively shortens the synthesis and potentially allows the application of this method to a broad scope. The only drawback of this method resides in the slight instability of aryl boron trifluorides that are partially hydrolyzed during the reaction, giving rise to the formation of the corresponding boronic acid. However, in this case, the side product is well defined from the targeted one and can be easily removed by preparative HPLC.
Sulforhodamine 6G was radiolabeled on an alkyl trifluoroborate group, in a similar way [
85]. The corresponding dual mode probe was obtained with a 25% d.c. RCY in 25 min and a A
m of 4 Ci/μmol. A trivalent version of the prosthetic group was also developed to synthesize probes bearing one sulforhodamine 6G and two RGD peptides with RCYs from 11% to 33% and A
m up to 3.95 Ci/μmol [
85,
86]. Up to now, only one fluorescein derivative has been synthesized by Ting’s group, who used the same technology [
87,
88]. A fluorescein derivative bearing an azide group was coupled to alkyl BF
3 moiety by Huisgen cycloaddition to afford the radiolabeling precursor. IEX reaction using similar conditions was then performed to obtain the corresponding PET/OPI dual probe with a 57% RCY in around 60 min and A
m 0.4 Ci/μmol from 116 mCi of [
18F]fluoride.
[
18F]Sulforhodamine B was also afforded by fluoride capture [
82]. Instead of a one-step radiofluorination from an arylboronic ester, a one-pot two-step starting from an arylborimidine bearing a terminal alkyne was chosen. Radiofluorination of the prosthetic group was performed by reacting
18F fluoride in acidic condition (pH = 2) in both non-carrier-added (NCA) and near-NCA conditions followed by a cycloaddition reaction to the azido-sulforhodamine B. In NCA conditions, the target compound was obtained with a 15% RCY and a very high A
m of 15 Ci/μmol, and near NCA conditions afforded a higher 30% RCY while allowing a lower but still high A
m of 7.5 Ci/μmol from around 20 mCi each (after trap** and concentration). Compared to the IEX-based methods, high molar activities are here allowed by the stoichiometry of the reaction, enabling the introduction of 3 F atoms, thus theoretically multiplying the A
m values by 3. Again, aqueous medium, mild conditions (30 °C), short reaction time (15 min), and use of non-dried
18F fluoride are quite appreciable conditions for radiolabeling of biomolecules.
Finally, AlJammaz et al. described the radiolabeling of a rhodamine conjugated with [
18F]2-fluoro-2-deoxyglucose ([
18F]FDG) [
89]. Radiosynthesis was performed quite easily by heating the precursor with [
18F]FDG in acetate buffer at 60 °C for 10 min. Reaction of the hydroxylamine with the aldehyde of the opened form of [
18F]FDG afforded the aldoxime conjugate with a nearly quantitative RCY (starting from [
18F]FDG) in around 20 min and a A
m of 70 mCi/μmol.
Xanthene derivatives proved to be a suitable platform for radiofluorination, affording dual mode PET/OPI compounds in good yields and high molar activities via various methods. However, emission maxima of current radiolabeled compounds do not exceed 590 nm, which could appear as a drawback compared to red-shifted dye such as Cy7. Moreover, radiolabeling sites are currently restricted to the central group, which could be a limitation in terms of synthetic approaches. Nevertheless, rhodamines’ outstanding optical properties make of them a dye of choice for the development of PET/OPI dual mode probes.
2.3. Cyanines
Cyanines are among the most used fluorophores along with BODIPYs and xanthene derivatives, especially for biomedical applications. First synthesized in 1856 by Williams, several types of cyanines have now been developed, including classical cyanines, hemicyanines, squaraines, merocyanines, and oxonols [
90,
91,
92]. To our knowledge, only classical cyanines have been labeled with
18F for dual-mode OPI-PET imaging. From a photophysical point of view, these dyes have several advantages such as high extinction coefficients and quantum yields in solution and can offer a red-shifted emission up to 1000 nm [
91,
93]. Cyanines display low toxicity and therefore are commonly used in biomedical applications. Notably, indocyanine green (ICG) and fluorescein are among the only fluorescent indicators approved by the Food and Drug Administration (FDA) and European Medical Agency (EMA), and IRDye800 CW is the subject of several clinical trials [
13,
94,
95]. Possible functionalization on terminal nitrogen atoms and on the central methine group is favored for introduction of bioconjugation links and radiolabeling sites. The weaknesses of cyanines for molecular imaging are mainly due to their low Δ
SS and their poor photostability, causing reabsorption and photo-bleaching, respectively. Since 2010, numerous ways of
18F radiolabeling cyanines have been described (
Scheme 4). Four pathways can be differentiated: late-stage fluorination by IEX, fluoride capture by boronic esters or aromatic nucleophilic substitution, and two-step labeling by introduction of a pre-labeled prosthetic group.
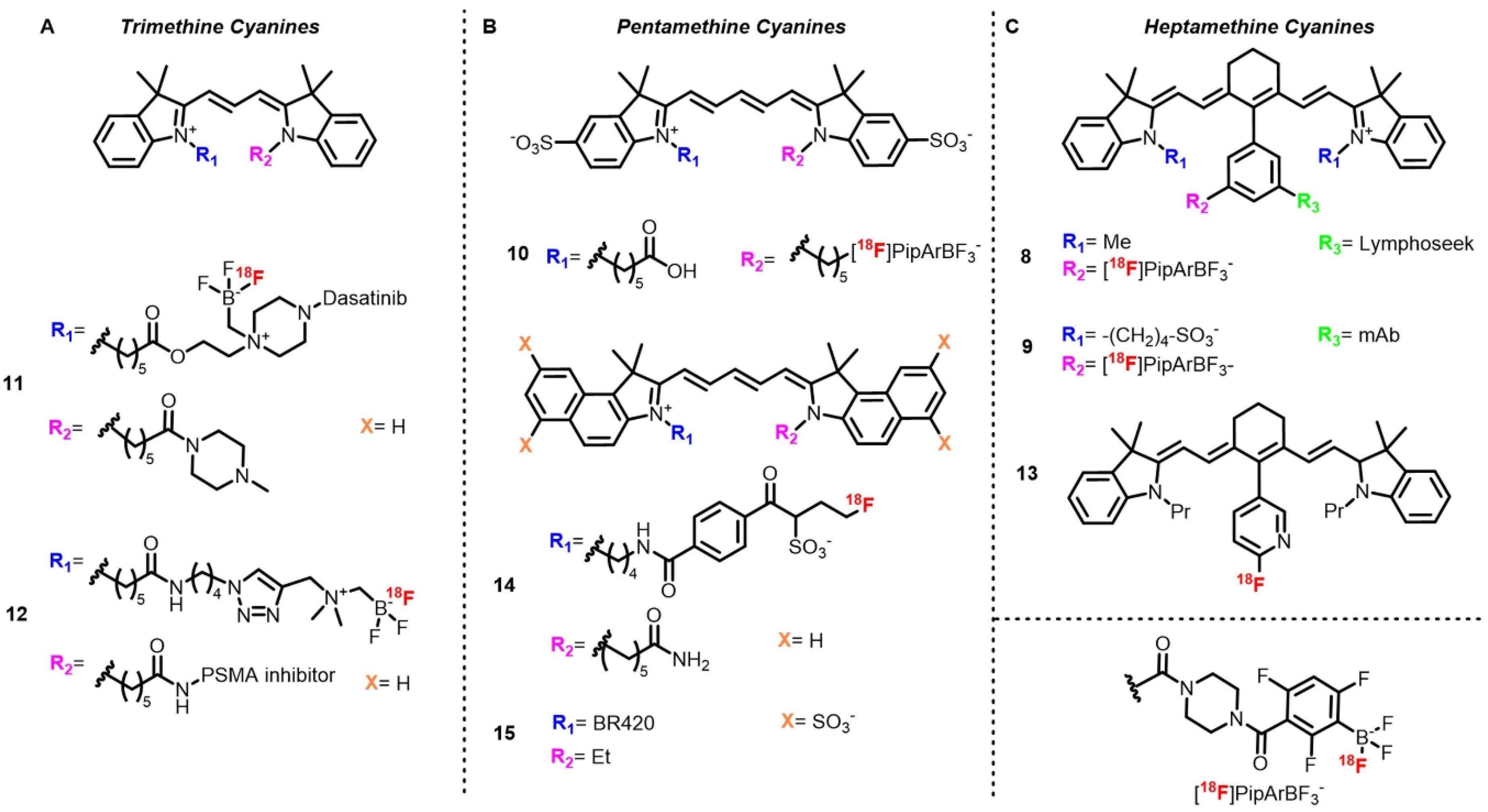
The first example describes
18F labeling of a heptamethine cyanine (Cy7) conjugated with Lymphoseek (
8), a dextran functionalized with mannose and diethylenetriamine penta-acetic acid (DTPA), used for lymph node imaging in melanoma or breast cancer [
43]. The method used relies on fluoride capture by a boronic ester in acidic aqueous conditions to afford a radiolabeled trifluoroborate [
58]. Although this method requires carrier-added conditions (KHF
2) and does not always allow separation of the radiolabeled trifluoroborate and the boronic ester, the first studies on this method described an A
m (d.c.) of 0.12 Ci/μmol from 100 mCi of starting activity. Radiolabeling of the Cy7–Lymphoseek conjugate following an optimized method afforded the target compound with RCY up to 3% and A
m values of 0.242 Ci/μmol. This procedure is quite appreciable for biomolecule radiolabeling as it is carried out in aqueous solutions and at low temperature. Moreover, starting activity is used as received at the end of the beam without a drying step. In this case, low A
m did not prevent imaging experiments, as the authors determined that A
m from 1 to 50 Ci/mmol was sufficient. Ting’s group also described a late stage radiolabeling of Cy7–mAb conjugate (
9) (cetuximab) by adaptation of this method to solid phase synthesis [
96]. The authors took advantage of the presence of the dioxaborolane group to introduce a biotin handle used for immobilization on an agarose-streptavidin gel. Once treated with [
18F]fluoride, the target radiotracer, which was no longer bound the gel and possessed A
m = 0.16 Ci/μmol after purification by SEC-HPLC, was able to be injected directly after elution. The same reaction was utilized for a
18F N-functionalized pentamethine sulfo-cyanine pentamethine (Cy5) with a free carboxylic acid (
10) moiety for tumor xenograft imaging [
97].
IEX was also described on cyanine-based dual mode probes by Ting and coworkers, who labeled a trimethine cyanine (Cy3) functionalized on one of the nitrogen atoms with dasatinib (
11), a kinase inhibitor used for glioma treatment and imaging [
98]. Last step radiofluorination was then performed by heating the precursor with [
18F]fluoride in acidic conditions. Attractively, this method is simple, rapid, and withstands water, as no quaternary methyl ammonium (QMA) trap** and azeotropic drying are required. Purification is performed by reverse phase HPLC, with the probe being obtained in 62% d.c. RCY with A
m = 0.38 Ci/μmol in 26 min. The same method was used to label a sulfocyanine Cy3–PSMA conjugate (
12) where the alkyl BF
3 group was attached on one terminal nitrogen and PSMA on the other nitrogen atom [
99,
100]. The probe was obtained in lesser yield but comparable A
m, likely due to the heating (10–15 min at 80–90 °C). This tracer was purified by slow elution over a C18 cartridge with the help of a syringe pump. The authors emphasized the importance of the slow flow rate (40 mL/h) to avoid coelution with unreacted [
18F]fluoride.
Recently, Valliant et al. described introduction of
18F directly at the Cy scaffold by aromatic nucleophilic (S
NAr) substitution on a 2-nitropyridine group attached on the central methine position (
13) [
101]. In this example, the precursor used was a reduced cyanine (hydrocyanine) used as a turn-on optical probe upon oxidation for reactive oxygen species (ROS) imaging. The target tracer was obtained by usual S
NAr conditions by heating in presence of [
18F]KF and K
222 in basic polar conditions followed by preparative HPLC purification. If this method works quite well in this case, late-stage nucleophilic substitutions are usually limited to small molecules labeling or performed on prosthetic groups prior to bioconjugation due to the prolonged heating required.
Indirect radiolabeling of cyanine dyes have also been described. The first example was published by Priem et al., who developed a hydrophilic prosthetic group on the basis of the ring opening of a propanesultone with fluoride and bearing a conjugation moiety [
102]. Preparation of the prosthetic group was performed in three steps in 75 min, with an overall RCY of 20–30% d.c. before ligation to Cy5.5 via an NHS ester (
14). Although the synthesis of the prosthetic group seems tedious and long, it represents a potential way to modulate the pharmacokinetics of small radiotracers by improving their hydrophilicities thanks to the sulfonic acid group. Moreover, very quick coupling reaction (1 min) and purification (SPE) shortens the overall synthesis. Another way to tune the pharmacokinetic of cyanine-based dual-mode tracers has been described by Schwegmann et al., who used mono-, bis‑, and tetra-sulfonated Cy5 coupled to BR420 (
15), a barbiturate-derived inhibitor, to favor renal over hepato-biliary clearance [
103]. Radiolabeling was performed by click chemistry ligation of [
18F]
1-azido-2-fluoroethane and the set of radiotracers were obtained in 104 to 138 min, with RCY between 9.7 and 19.2% d.c. and A
m ranging from 0.03 to 0.97 Ci/μmol.
In the view of dual mode PET/OPI probes, the major advantage of Cy dyes is the facile access of near-infrared emitting dyes, as commercially available Cy dyes can emit up to 800 nm. However, as Schwegmann et al. observed, non-hydrophilic Cy dyes are quickly cleared via the hepatobiliary system and might need to be replaced with sulfo-cyanines.
2.4. Curcumins
Curcumin is the major pigment of turmeric roots and a component of eastern traditional medicine and gastronomy [
104]. It has long been studied for its biological and photophysical properties as it displays anti-cancer and neurological activities, among others [
105,
106]. As a fluorophore, curcumin exhibits only modest characteristics with absorption and emission bands located around 420 and 500 nm, respectively; Φ
F up to 17% in low polarity media; and fluorescence lifetimes below the nanosecond [
104]. Moreover, its bioavailability is rather low due to poor hydrolytic stability [
104,
107]. However, its interesting biological properties have still encouraged researchers to radiolabel curcumin derivatives for amyloid plaques and cancer imaging. Moreover, complexation to a metal center and engineering on curcumin scaffold were performed in order to improve its properties and allow its use as a PET/OPI dual mode probe [
107,
108,
109]. Radiofluorination of curcumin derivatives is mostly performed on the phenol groups by introduction of a
18F fluoroalkyl or fluoroethyleneglycol chain. It can also be labeled on the polymethine chain after modification of the β-diketone moiety.
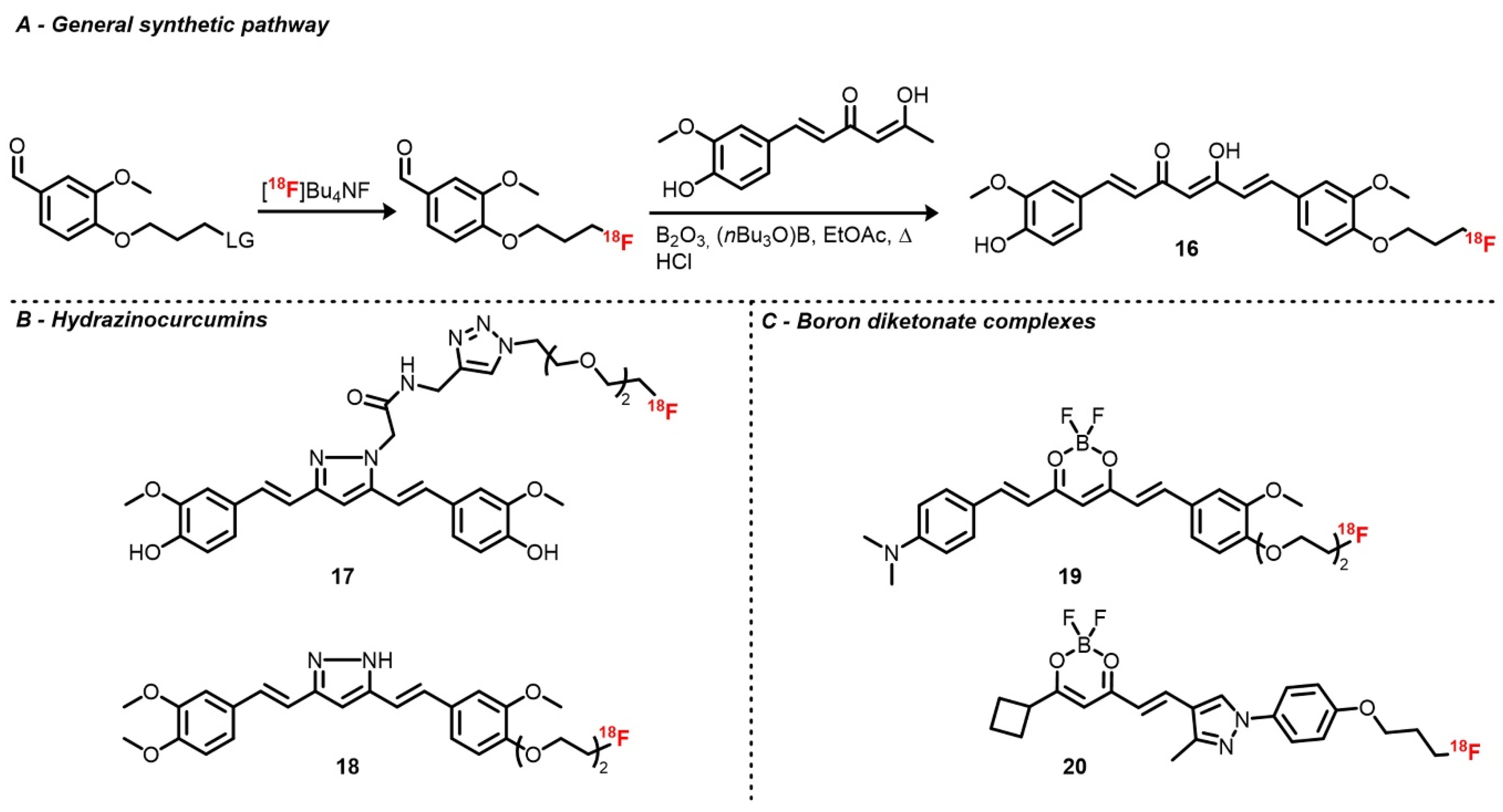
Ryu et al. investigated two curcuminoid radiofluorination methods [
110]. First, relying on the classical nucleophilic substitution of a tosylate by [
18F]fluoride on a curcumin derivative functionalized with a propyltosylate group on one of the two phenols, the corresponding [
18F]fluoropropylcurcumin (
16) was afforded with poor RCY and A
m, likely due to curcumin instability. As an alternative, [
18F]4-fluoropropyl-2-methoxybenzaldehyde was synthesized the same way prior to an aldolization step (105 °C for 1 min in EtOAc) with the boron diketonate complex, which was eventually hydrolyzed (
Scheme 5). After HPLC purification, [
18F]fluoropropylcurcumin was obtained with d.c. RCYs of 16–25% and A
m = 1.01 Ci/μmol. As brain uptake of this tracer was slightly lower than expected, an analog where the second hydroxy group was replaced by a methoxy was synthesized in order to improve its lipophilicity. The fluoropropyl chain was also replaced by a fluoroethyl one [
111]. Along similar lines, curcumin derivatives with a pyrazole ring instead of the 1,3-diketone were developed. Rokka et al. described the radiolabeling of a pyrazole curcumin analog bearing a linker with a terminal alkyne aimed for prosthetic group introduction via cycloaddition [
112]. This precursor was radiofluorinated with a one-pot two-step procedure by heating [
18F]potassium fluoride–Kryptofix complex with azido diethyleneglycol tosylate in ACN, followed by cycloaddition at room temperature in the presence of copper sulfate. The radiotracer (
17) was obtained with an overall RCY of 21 ± 11% and a very high A
m, which was above 27 Ci/μmol. Shin et al. also described the radiofluorination of hydrazinocurcumin (
18) analog with a diethyleneglycol tosylate chain on one of the two hydroxy groups [
113]. Protection of the remaining hydroxy function and of the pyrazole NH group with methoxy methyl ether (MOM) was necessary to afford proper yields. Radiotracers were then obtained by classical substitution followed by acidic deprotection of MOM groups with d.c. RCY from 25 to 35% and A
m around 1.3 Ci/μmol.
Recently,
18F boron diketonate complexes were also described following the one-pot two-step method initially described by Ryu et al. [
114,
115]. Almost simultaneously, the groups of Seong Choe and Ran published radiofluorinated BF
2 complexes of curcumin (
19) and half-curcumin derivatives (
20), respectively. Both probes were inspired by the CRANAD family, a group of fluorescent dyes based on the BF
2 diketonate scaffold [
108]. Tracers
19 and
20 were obtained with d.c. RCYs around 20% and 51% and A
m of 1.16 and 1.19 Ci/μmol, respectively. During the development of these tracers, Kim et al. investigated the acid-mediated IEX radiofluorination in a similar manner as for BODIPYs, however, it was unsuccessful and only decomposition occurred [
114]. Recently, Ting’s group patented a radiofluorinated curcumin derivative labeled via IEX using SnCl
4 [
116].
Although curcumin-based probes display limited optical and biological properties compared to the aforementioned scaffolds, the work described thus far paves the way for the development of PET/OPI probes, especially for amyloid plaque imaging. Efforts to improve curcumin’s in vivo stability should also be maintained in order to reach clinical translation.
2.5. Others
Aside from BODIPYs, cyanines, and xanthenes, many other fluorescent scaffolds have been labeled with 18F. However, use of their optical properties is mostly restricted to cell imaging.
In some cases, fluorescent scaffolds are introduced not in light of optical imaging experiments, but rather because they display good affinities with biological targets. This case is well illustrated by coumarins, which are common fluorophores. Up to now,
18F-labeled coumarins have only been used for their affinities with diverse targets such as dopamine D4 receptor and carbonic anhydrase IX [
117,
118,
119,
120].
18F was introduced either via substitution on an alkyl tosylate chain or by S
NAr on prosthetic groups affording tracer in low to moderate RCYs and good A
m. The absence of optical studies is likely due to the modest optical properties of the native backbone (blue centered absorption and emission, low Φ
F) compared to fluorophores discussed above. However, after suitable functionalization, i.e., extension of the π-delocalized path or introduction of an electron-donating group, development of dual-mode PET/OPI probes could be considered [
121].
A few examples of radiofluorinated porphyrinoids have also been described. Most recent syntheses use either classical direct tosylate substitution or click ligation with the precursor in decent yields and molar activities [
122,
123,
124]. Kavali et al. also described condensation of [
18F]
p-fluorobenzaldehyde with pyrrole and
p-methoxybenzaldehyde or on the corresponding tetrapyrrane precursor [
125]. Although existing works focus mostly on radiosynthesis and preclinical evaluation based on PET and cellular uptake, use of the optical properties of porphyrin derivatives in OPI and especially photodynamic therapy (PDT) are promising. However, compared to their radiometalated analogs,
18F-porphyrinoids are still seldomly used, probably due to the shorter half-life of
18F, which sets time limitations in terms of synthesis and analysis. Moreover, introduction of
18F requires design of the precursor as it contains a labeling function that is not required in radiometallated porphyrins.
Well-represented
18F tracers displaying fluorescence properties include azo dyes, stilbenes, phenazine derivatives, and 2-(aryl)azoles or benzazoles [
126,
127]. These scaffolds are often met in brain imaging projects but their optical properties (low ε, Φ
F in solution) often limit their use to conventional PET tracer evaluation and tissue section imaging. This is also the case for compounds that are not part of usual dye families [
128,
129].
18F-labeled analogs of common staining dyes have also been described. Indeed, acridine orange, ethidium, anthracene, and Evans blue analogs have been radiofluorinated, but only [
18F]ethidium optical properties were used in a cell imaging experiment despite their red-centered emission [
130,
131,
132,
133,
134].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






