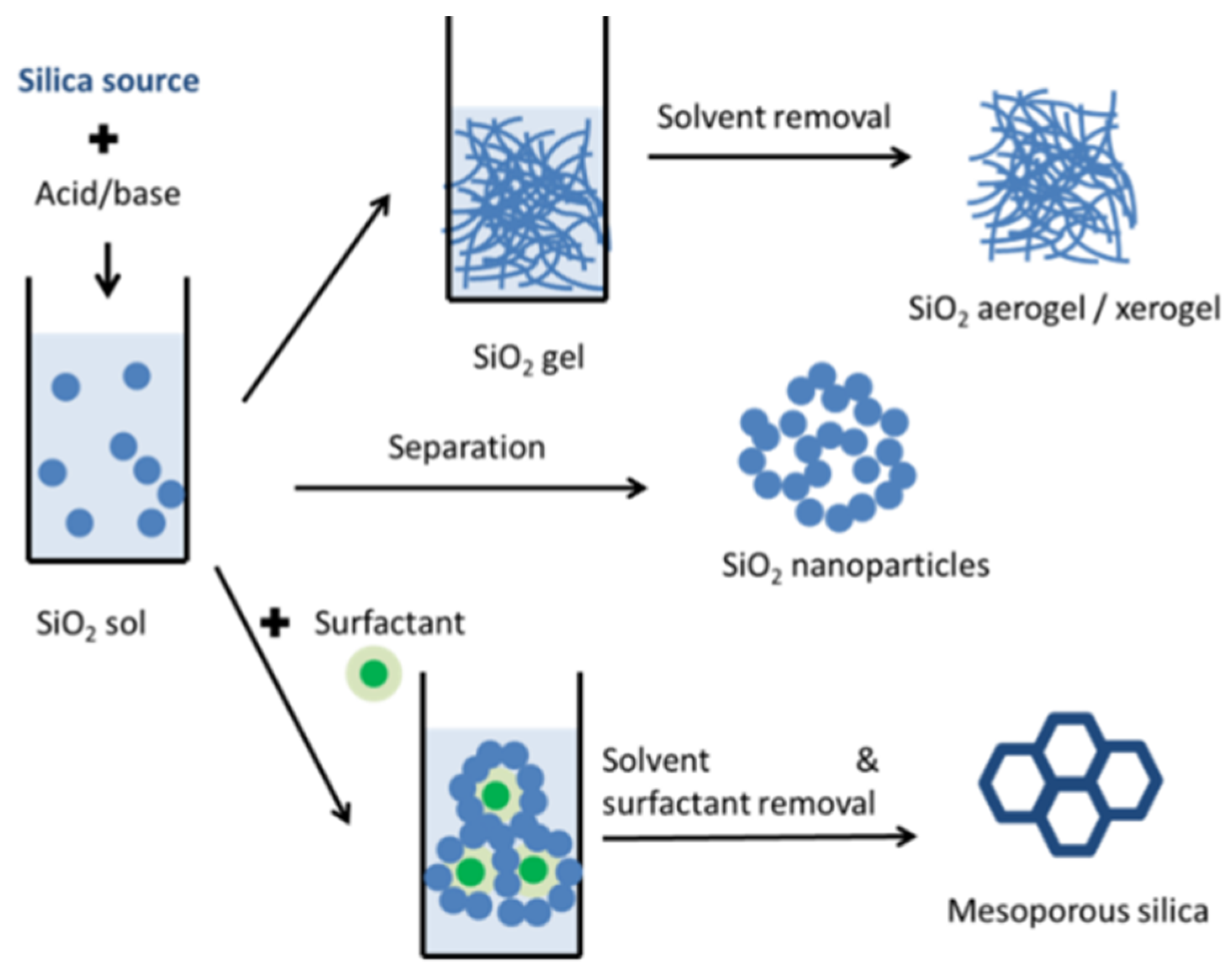
The main organic phase change materials consist of paraffins, fatty acid, and their derivatives, sugar alcohols and polymers, while the principal inorganic PCMs are salt hydrates, molten salts, and elemental compounds. The most used porous silica matrices include aerogels and xerogels, mesoporous silica and silicates, silica nanoparticles either as-prepared or fused. A large variety of possible nanocomposites can be obtained, enabling control over the thermal and structural properties. Additional materials are sometimes added to improve the properties of the resulting samples. The most common additives are carbon-based materials, which have high thermal conductivity. The recent progress in the field of porous silica-based phase change materials is presented in the following subchapters, based on the main type of PCM used to obtain the nanocomposites.
3.1. Paraffins
Paraffins or paraffin waxes are mainly composed of long chained n-alkanes. Although flammable, they are relatively unreactive. While pure compounds are sometimes used, most often paraffins are mixtures. A high number of paraffins are commercially available, covering a large temperature range in terms of their melting points. They also have high heat of fusion values, often exceeding 200 J g−1. The long alkyl chains have a parallel arrangement in solid state, which requires a high amount of energy to break, thus yielding high heat of fusion values. N-alkanes sometimes exhibit a secondary solid-solid phase transition below their melting point. This is an order-disorder transition, caused by a partial destruction of the parallel chain structure. Paraffins are a by-product of petroleum refining, making then attractive from the standpoint of waste reutilization.
Paraffin–porous silica composites can be obtained through direct sol-gel synthesis of the silica matrix in the presence of paraffin, a suitable structure directing agent, and sometimes an emulsifier [
82,
83,
84,
85]. Cetyltrimethyl ammonium bromide (CTAB) and
n-pentanol as emulsifier were found to give superior results over sodium dodecyl sulfate (SDS) or Span 80 and Tween 80 [
82]. A composite with a heat storage capacity of 95 J g
−1 and a m.p. of 30 °C was obtained when starting from a paraffin wax with a Δ
Hf value of 142 J g
−1 and similar m.p (
Table 3). Simple hydrolysis of tetraethyl orthosilicate in acid medium could yield a composite with up to 92% wt. paraffin, although it is unlikely such a material also had shape-stability [
83]. Similarly, paraffin with 110 °C m.p. was encapsulated into silica at a 1:1 wt. ratio of paraffin to TEOS [
84]. The material was added in-situ for providing thermal control of the methyl methacrylate polymerization reaction. An optimization of the sol-gel synthesis using sodium silicate in terms of pH, temperature, paraffin/water and paraffin/silica was carried out [
86,
87]. Shape-stabilized composites could be obtained at pH = 4.5 and 35 °C. Nanoencapsulated
n-octadecane into 170–560 nm silica shells shows a remarkable dependence of the melting point decrease with particle size [
88]. The m.p. decreased varies from 2.2 °C for the 170 nm nanoparticles to 1.2 °C for the 560 silica NPs. C17-C20
n-alkanes, sodium silicate and Pluronic P104 were used to create shape-stabilized PCMs through direct sol-gel synthesis [
89]. Around 45–55% wt.
n-alkanes were encapsulated, while the composite melting enthalpy varied between 61–81 J g
−1 depending on the hydrocarbon chain length. The composites had increased thermal stability with respect to pure paraffins and good reliability after 100 heating-cooling cycles.
Liu et al. designed mesoporous silica nanospheres and nanocapsules for
n-eicosane through direct sol-gel synthesis [
90]. The matrices have pore diameters of 7.9 and 3.3 nm and could be used to obtain shape-stabilized PCMs with 122 and 113 J g
−1 heat of fusion values for the nanospheres and nanocapsules, respectively. The thermal conductivity increased from 0.15 for the pure paraffin to 1.17 W m
−1 K
−1 for the nanocapsules.
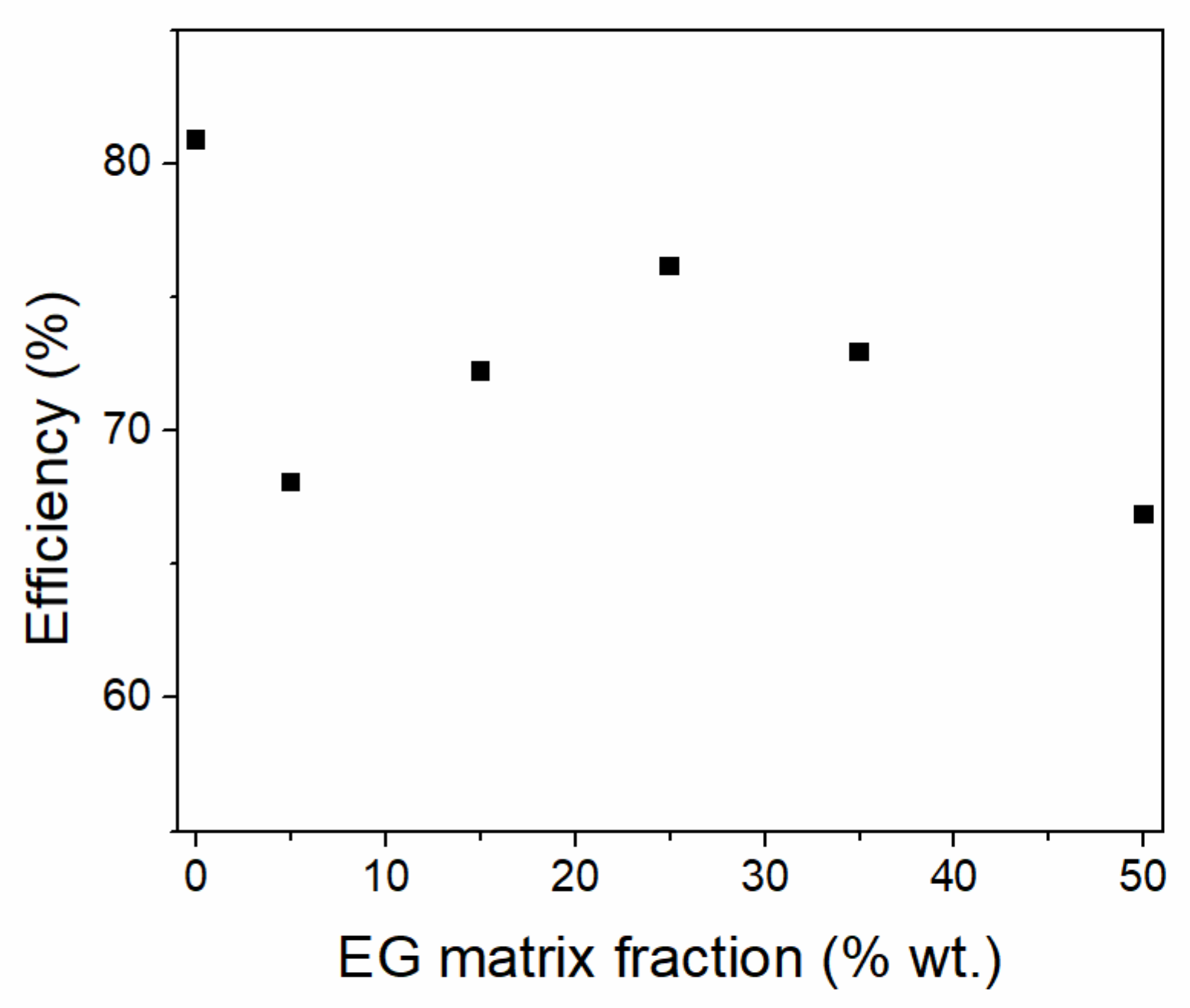
A comparison of the initial amount of various PCMs in the one-pot sol-gel synthesis of silica PCMs was carried out using paraffin, stearic acid and polyethylene glycol (PEG) at 30, 50 and 60% wt. ratios [
85]. No heat storage capacity was obtained for any PEG-based PCMs, while a significant loss of thermal energy storage at low PCM ratios, especially for the stearic acid was noticed (
Figure 6). The reduction in heat storage was correlated with the presence of nanoconfined PCMs into the silica mesopores. The thermal energy efficiency can be computed as the ratio of effective PCM content calculated based on the experimental heat of fusion values divided by the actual PCM content (
Figure 6).
Impregnation of molten paraffins into an already prepared porous matrix is also used to create shape-stabilized PCMs [
27,
91,
92,
93,
94]. This method offers better control over the silica pore shape, size and volume as well as paraffin mass fraction, at the cost of a two-step synthesis. Porous silica aerogels obtained by supercritical ethanol extraction were impregnated with a paraffin with a 56–58 °C melting range [
27]. The most porous aerogel, with an average pore diameter of 56 nm and total pore volume of 5.22 cm
3 g
−1, yielded a composite with 75% paraffin. Vacuum melt impregnation was used to create expanded perlite-paraffin shape-stabilized composites, which were then coated with colloidal silica and acrylic and used to create energy storage panels for buildings [
91]. Larger, 1 mm expanded perlite particles were found to have lower leakage amounts than smaller, 60 μm particles in a later study [
95]. Paraffin with 28 °C m.p. was melt impregnated into raw diatomite and mesoporous MCM-41 silica [
92]. Up to 55% and 60% wt. heat storage compound could be impregnated into diatomite and MCM-41, respectively. The m.p. was decreased with 2.5–3.5 °C with respect to bulk, indicating interparticle nanoconfinement effects. Vacuum melt impregnation into a silica aerogel-polytetrafluoroetheylene (PTFE) thin film achieved 62.8% wt. paraffin loading [
96]. The experimental heat of fusion value was lower than the expected value (η = 83.9%), indicating the formation of a non-melting layer or an amorphous phase. The optical transmittance of the PCM thin film exhibited both temperature and wavelength dependence. Fumed silica-paraffin nanocomposites were added up to 30% wt. into cement formulations for building passive energy storage applications [
93]. A shape-stabilized paraffin/fumed silica sample was obtained at 45% wt. loading and it exhibit 100% heat storage efficiency. Increasing the mass fraction of the composite PCM into the cement mixture led to a decrease of thermal conductivity from 0.127 W m
−1 K
−1 to 0.101 W m
−1 K
−1 at 30% wt. composite. Further studies showed that the addition of 3% silica nanoparticles and PCMs (paraffin or
n-octadecane) to cement mixtures led to better mechanical properties and low chemical shrinkage [
97]. A kinetic study of paraffin melt impregnation into silica aerogel showed that a maximum loading of 75% wt. can be achieved after 3 min, with little further improvements [
94]. Melt impregnation of octadecane and a complex mixture of fatty acids into micronized silica nanoparticles have resulted in composites with similar melting points and reduced heat storage efficiencies [
98].
A comparative study of vacuum melt impregnation into mesoporous silica, expanded graphite (EG), bentonite, diatomaceous earth and zeolite Y was carried out using hexadecane, octadecane, capric and lauric acid eutectic mixture and butyl stearate [
99]. The highest hexadecane loading was obtained in the case of the mesoporous silica matrix, at 81% wt. EG exhibited PCM loading of 77% wt., with the rest of the matrices loading less than 50% PCM. The reliability of the best two matrices was tested up to 450 heating–cooling cycles at three initial PCM to matrix rates. It was found that all samples stabilize after 300 cycles. Heat of fusion vales around 100 J g
−1 could be obtained for starting PCM: matrix ratio of 2:1 and 3:1. Nomura et al. studied the influence of varying the pore diameter of mesoporous silica between 11 and 50 nm on the thermophysical properties of the resulting octadecane-silica nanocomposites [
81]. The authors found that pore radius more than 20 nm are required for the nanocomposites to retain at least 80% of their theoretical heat of fusion. Decreasing pore diameters below 40 nm leads to fast reduction in heat storage due to the increased volume occupied by the non-melting layer, as well as a proportional reduction of m.p. following the Gibbs-Thompson equation.
Solution impregnation methods can also be used to obtain paraffin–porous silica nanocomposites [
100]. For example, ASTM D 87 paraffin wax was impregnated into two types of Stöber silica nanoparticles from a 1:1 (
v/
v) ethanol/hexane solution in the presence of polyvinyl pyrrolidone (PVP) at different mass fractions [
100]. Significant paraffin loading was achieved only when using more than a 2:1 wt. paraffin to silica ratio in the initial synthesis. No supercooling was noticed, while a slight decrease in heat of fusion values indicated the presence of a non-melting layer.
A comparative study of melt impregnation, solution adsorption followed by filtration or evaporation was carried out using
n-eicosane and silica aerogel (Enova aerogel particles IC3120, Cabot Corporation), with heptane as the solvent [
101]. The highest eicosane adsorption of 84.3% wt. was obtained for melt impregnation, with the solvent methods yielding 72–75% wt. paraffin loadings. Thermal energy storage was proportional to the paraffin mass loading. The particles could be used for coating textiles to provide thermal comfort in the 35–39 °C temperature range.
The hydrophobic nature of the paraffins contrasts with the hydrophilic silica surface containing surface silanol (Si-OH) groups. One of the earliest examples in modifying the silica surface with hydrophobic moieties is the study of Li et al., which used hydrophobic fumed silica to decrease paraffin leakage from a 30:70 wt. PCM: cement formulation [
102]. Leakage was eliminated when 9% wt. hydrophobic fumed silica was used with respect to pure paraffin. A follow-up study also included hydrophilic and hydrophobic silica aerogels and commercial RT21 paraffin [
103]. The hydrophilic and hydrophobic silica aerogels were found to possess higher adsorption capacity for the paraffin, at 78 and 75% wt., respectively. The lower adsorption capacity of the hydrophobic aerogel could be caused by lower pore volume due to its modification. Flexible monolithic aerogels functionalized with methyl groups were obtained and used for paraffin impregnation from ethanol solution followed by evaporation [
104]. Shape-stabilized samples were obtained at 70% wt. paraffin loading. The presence of a non-melting layer was assessed by the lower heat storage efficiency of the prepared composite. Similarly, octadecane, octadecanol, and stearic acid were also impregnated at 70% wt. loadings and shown to yield shape-stabilized composites with high heat of fusion values, between 127 and 141 J g
−1 [
104]. A mesoporous silica aerogel was obtained and functionalized post-synthesis through alkylation with methyl groups or by hydroxylation [
105]. Paraffin wax was loaded through vacuum melt impregnation. The paraffin mass fraction was dependent on both pore volume and surface functionalization. Pore filling values below 100% were noticed for hydroxyl groups, while complete filling was achieved for methyl functionalization. Interestingly, a value of 168% pore filling was obtained for dual methyl and hydroxyl functionalization, at a paraffin loading of 88% wt., indicating adsorption in interparticle spaces [
105]. A comparative study of hydrophilic or methyl-functionalized aerogel melt impregnated with paraffin RT60 was also carried out [
106]. The methyl-functionalized aerogel yielded composites containing 80% wt. paraffin and 180 J g
−1 heat storage capacities. A hierarchically porous silica monolith was functionalized with alkyl chains of varying length and used to adsorb octadecane and stearic acid [
107]. The functionalized matrix presented a nanoconfined phase melting with higher enthalpy for the octadecyl functionalization than for octyl moieties. This small increase could be explained by cooperative interactions between the alkyl chains of the PCM and the silica surface functionalization, especially if both had the same length (e.g., C18 chains). Dodecane was adsorbed at 75 and 85% wt. loading into hydrophobic fumed silica [
33]. The composite containing 75% paraffin exhibited shape-stabilization, while some leakage was noticed at higher loadings. On the other hand, the thermal conductivity increased proportional to dodecane loading. A similar result was obtained using RT28 paraffin and hydrophobic fumed silica for Li-ion battery thermal protection applications [
108]. While composites with 70% paraffin exhibited shape-stability, leakage was noticed at 80% wt. loading.
Gas phase synthesis of composite PCMs was carried out by Choi et al., using mesoporous SBA-15 matrices with hexagonally ordered cylindrical mesopores [
109]. The solid PCM was heated under vacuum in the presence of the silica matrices having pore diameters ranging from 5.6 to 12.5 nm. Octadecane and tetradecane were used as paraffins, while dodecanoic acid, decanoic acid, tetradecanol, and dodecanol were also employed. The pore fill ratio decreased from ~100% for the 5.6 nm SBA-15 to 65% for the 12.5 nm silica matrix. The authors presented evidence for both a decrease of the nanoconfined PCM melting point following the Gibbs-Thompson equation as well as for the existence of the non-melting interface layer, which caused a decrease of the recovered heat of fusion and better fit for the nanoconfined m.p. decrease (
Figure 7). A subsequent study used docosane (C24
n-alkane) impregnated by the same method into SBA-15 mesoporous silica, resulting in a composite PCM with 53.3 J g
−1 heat of fusion at 31 °C. [
110] The composite was then theoretically investigated for dehumidifying in a fixed-bed adsorption process and it was found to have high performance due to the ability to recover heat during the first 5 min of the process.
The addition of carbon-based materials has been investigated for increasing the thermal conductivity of porous silica–paraffin composites [
111]. Expanded graphite (EG)-silica-paraffin composites were obtained through one-pot sol-gel synthesis [
111]. The composite had double the pure paraffin thermal conductivity, as well as high (104.4 J g
−1) heat storage capacity at 27.7 °C. The influence of paraffin mass fraction was investigated in a latter work. Composites containing 2% expanded graphite, 20 nm silica nanoparticles and between 86–92% wt. paraffin were obtained through the addition of the components to melted paraffin [
112]. All samples containing at most 90% wt. paraffin presented shape stability. The thermal stability, conductivity, and drop point all increased with decreasing paraffin content. Low-density polyethylene (LDPE) was also added during the sequential melt synthesis of an EG/SiO
2/LDPE/Paraffin composite containing 30% wt. LDPE, 7% wt. EG and 3–7% wt. silica nanoparticles [
113]. The 5.5% wt. silica composite showed the least leakage and was further tested for Li-ion battery thermal protection.
N-eicosane was loaded into 20 nm SiO
2 nanoparticles (NPs) from ethanol solution followed by evaporation, at different loadings between 65 and 75% wt. [
114]. Shape-stabilized composites were obtained up to 70% wt. loading. Additional samples containing 3, 5, to 7% wt. EG were similarly prepared at a 7:3 eicosane: silica mass ratio. The thermal conductivity increased proportionally with the EG fraction, being 2.3 higher than pristine eicosane at 7% wt. EG. Porous matrices were also developed from silica gel industrial waste, which was hydrolyzed to sodium silicate [
115]. Paraffin wax was vacuum melt impregnated at 80% wt. loading, while the matrix composition was varied between 0 and 50% wt. EG. The thermal conductivity of the composites increased linearly with EG content. All samples exhibit shape-stability and reliability after 1000 heating-cooling cycles [
115]. While the experimental heats of fusion are lower than the theoretical value, indicating the presence of a non-melting layer, there is no linear correlation between experimental heat of fusion and EG weight fraction (
Figure 8). It is worth noting that simulations of heptadecane encapsulated into MCM-41 have shown a two-fold increase in thermal conductivity over pristine paraffin, without the addition of carbon-based materials [
116]. Moreover, the thermal conductivity was shown to increase with increasing PCM amount up to 70%.
Fumed silica and graphite powders were used to encapsulate paraffins for the addition into cement [
117]. The obtained “thermocrete” had a latent heat storage capacity of 12.6 J g
−1, with a melting point of 32.9 °C and a freezing point of 17.1 °C.
Graphene oxide (GO) was added during the synthesis of a paraffin–silica composite with the goal of increasing thermal conductivity [
118]. The synthesis was carried out in the presence of polyvinyl alcohol (PVA) and mixed Span80 and Tween80 surfactants. The thermal conductivity of the composite is significantly increased to 1.16 W m
−1 K
−1 in comparison with pristine paraffin, at around 0.34 W m
−1 K
−1. A similar approach consisted of adding GO to molten paraffin in the presence of a surfactant followed by porous silica synthesis through interfacial polycondensation in an oil–water emulsion [
119]. Then, 5% wt. of the paraffin-GO-SiO
2 material was added to PVC formulations, which showed improved thermal conductivity with respect to pristine PVC.
Other functionalities can also be added to the composite PCMs. For example, magnetic response coupled with thermal storage was achieved by adding Fe
3O
4 nanoparticles to a
n-eicosane (C
20H
42) suspension, followed by silica hydrolysis and condensation in the presence of CTAB, which resulted in 4–6 μm core-shell particles [
120]. Materials with up to 70% wt. PCM versus SiO
2 were obtained, possessing the corresponding heat of fusion based on the paraffin weight fraction (
Table 3). Ti
4O
7 was added to paraffin-silica nanocomposites for solar energy adsorption [
121]. A 3.3-fold increase in thermal conductivity with respect to pristine paraffin was also achieved for the SiO
2-Ti
4O
7 matrix.
Table 3.
Representative porous silica–paraffin nanocomposites.
Table 3.
Representative porous silica–paraffin nanocomposites.
| PCM | Porous Silica Composite | Ref. |
|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Sample/Synthesis | %PCM (wt.) * | m.p. (°C) | ΔHf (J g−1) | |
|---|
| Paraffin wax | 29.0 | 142.0 | Direct synthesis/CTAB + n-pentanol | - | 30.0 | 95.0 | [82] |
| Paraffin | 51.0 | 151.5 | Direct synthesis/TEOS/HCl | 92.1 | 50.2 | 139.6 | [83] |
| Paraffin | 57.0 | - | Aerogel/supercritical EtOH | 75 | - | - | [27] |
| Rubitherm RT 28 | - | - | TEOS/HCl, EG, paraffin | - | 27.7 | 104.4 | [111] |
| n-eicosane | 39.2 | 237.1 | n-C20H42,Fe3O4@SiO2 | 70 | 39.2 | 170.2 | [120] |
| PX25 | ~23 | 96 | 9% hydrophobic fumed silica vs. PCM; 30:70 wt. composite: cement | 27.3 | ~23 | 14.2 | [102] |
| Paraffin | 50.1 | 173.9 | Parafin@SiO2-GO/PVA, Span80, Tween80 | 49 | 49.7 | 87.1 | [118] |
| 25# Paraffin | 25.8 | 107.6 | Vacuum melt impregnation | - | 21.6 | 56.3 | [91] |
| n-eicosane | 37.13 | 249.0 | Aerogel melt impregnation | 84.3 | 36.8 | 198.4 | [101] |
| Paraffin | 57.7 | 161.4 | TEOS/HCl | 60 | 58.2 | 98.0 | [85] |
| Paraffin | 51.9 | 184.1 | 2% EG; 8% 20 nm SiO2; melt addition | 90 | 51.8 | 168.3 | [112] |
| Octadecane | 26.5 | 2300 | Gas transport/12.5 nm SBA-15 | - | 17.4 | 103.3 | [109] |
| Tetradecane | 6.2 | 216.0 | - | −7.4 | 124.2 |
| Paraffin | 49.7 | 200.4 | Solvent impregnation/SiO2 NPs | 80 | 52.0 | 156.6 | [100] |
| Paraffin | 28.0 | 168.0 | Melt impregnation/MCM-41 | 60 | 25.5 | 95.0 | [92] |
| Paraffin | 42.2 | 243.0 | Vacuum melt/SiO2-PTFE aerogel | 62.8 | 42.0 | 128.0 | [96] |
| Paraffin | 59.6 | 191.1 | Solution impregnation/CH3-fucntionalized aerogel | 70 | 59.6 | 112.9 | [104] |
| n-Eicosane | 36.9 | 243.3 | Solution impregnation/7% EG/70:30 Eicosane:20 nm SiO2 NP | 65.1 | 35.4 | 135.8 | [114] |
| Paraffin wax | 63.8 | 209.1 | Vacuum melt/CH3/HO-aerogel | 88 | 63.7 | 163.6 | [105] |
| Paraffin wax | 25.7 | 198.0 | Vacuum melt/SiO2-EG 1:1 | 80 | 26.7 | 105.9 | [115] |
| Paraffin | 56.8 | 182.2 | Melt impregnation/aerogel | 75 | 56.3 | 165.2 | [94] |
| Hexadecane | 17.7 | 220 | Vacuum melt/mesoporous silica/300 heat-cool cycles | 45 | 17.1 | 100.1 | [99] |
| Octadecane | 29.9 | 223 | Vacuum melt/mesoporous silica | 45.3 | 28.9 | 84.5 | [81] |
| Octadecane | 28.5 | 212.6 | Direct synthesis | - | 26.3 | 99.3 | [88] |
| Nonadecane | 29.4 | 201.0 | Direct synthesis | - | 26.2 | 80.8 | [89] |
| Octadecane | 28.2 | 232.5 | Vacuum melt/SiO2 NP | 70 | 27.7 | 85.0 | [122] |
3.2. Fatty Acids and Derivatives
Fatty acids are naturally occurring carboxylic acids containing a long n-alkane or alkene chain. The high melting enthalpy of these compounds arises from the attractive interactions between the log chains as well as from hydrogen bonding between the carboxylic groups. Fatty acids are especially interesting as a source of renewable, green phase change materials. Similar compounds containing long alkane chains and hydrophilic groups (hydroxyl, amine, ester, amide etc.) can also be used as heat storage materials. Unlike paraffins, fatty acids and their derivatives present both hydrophobic and hydrophilic moieties, which enable additional pathways to tailor their adsorption and properties through functionalization of the porous silica matrices.
The direct sol-gel synthesis in the presence of fatty acids can be used to obtain porous silica composites with xerogel silica framework [
123]. Up to 75% wt. capric acid (CA)-palmitic acid (PA) eutectic yielded shape-stabilized PCMs through this method. While the composite exhibited 103.4 J g
−1 heat storage, its thermal conductivity was decreased. Exfoliated graphite nanoplatelets could be added up to 6% wt. in order to increase the thermal conductivity and simultaneously increase the fatty acid loading up to 85% wt. Up to 65% wt. lauric acid (LA) could also be encapsulated through direct synthesis [
124]. The composite m.p. was decreased by 2 °C with respect to bulk LA, indicating nanoconfinement effects. Careful selection of the synthesis method can result in the formation of core-shell particles. For example, PA microcapsules were obtained using SDS as surfactant [
125]. The acid m.p. was decreased by 1–2 °C with respect to bulk, while up to 89% heat of fusion versus bulk was obtained. A 72/28 capric-myristic acid eutectic was encapsulated into silica through direct sol-gel synthesis [
126]. The composite containing 40% fatty acids maintained its enthalpy and m.p. after 200 heating-cooling cycles. The direct sol-gel synthesis of stearic acid (SA)-silica composites showed increased leakage and mass loss for samples containing more than 76% wt. SA [
127]. Interestingly, all samples had lower m.p. and enthalpy values than expected on the basis of SA mass, indicating the presence of a non-melting layer for this type of materials. Shape-stabilized PCMs containing up to 70% wt. 1, 8-octanediol (ODL) were also prepared by the sol-gel method [
128]. While the m.p. decreased by around 1 °C in comparison with bulk ODL, the enthalpy was close to the value expected based on the mass fraction of the diol (
Table 4).
The influence of methyl groups on lauric acid composite PCMs obtained through direct sol-gel synthesis was investigated using methyl triethoxysilane (MTES) as the organic group source [
129]. MTES to TEOS weight ratios of 0.45 to 0.53 yield composites with the best thermal energy storage properties as well as hydrophobicity (water contact angle of 114–125°) which could enable these materials to store heat in humid environments.
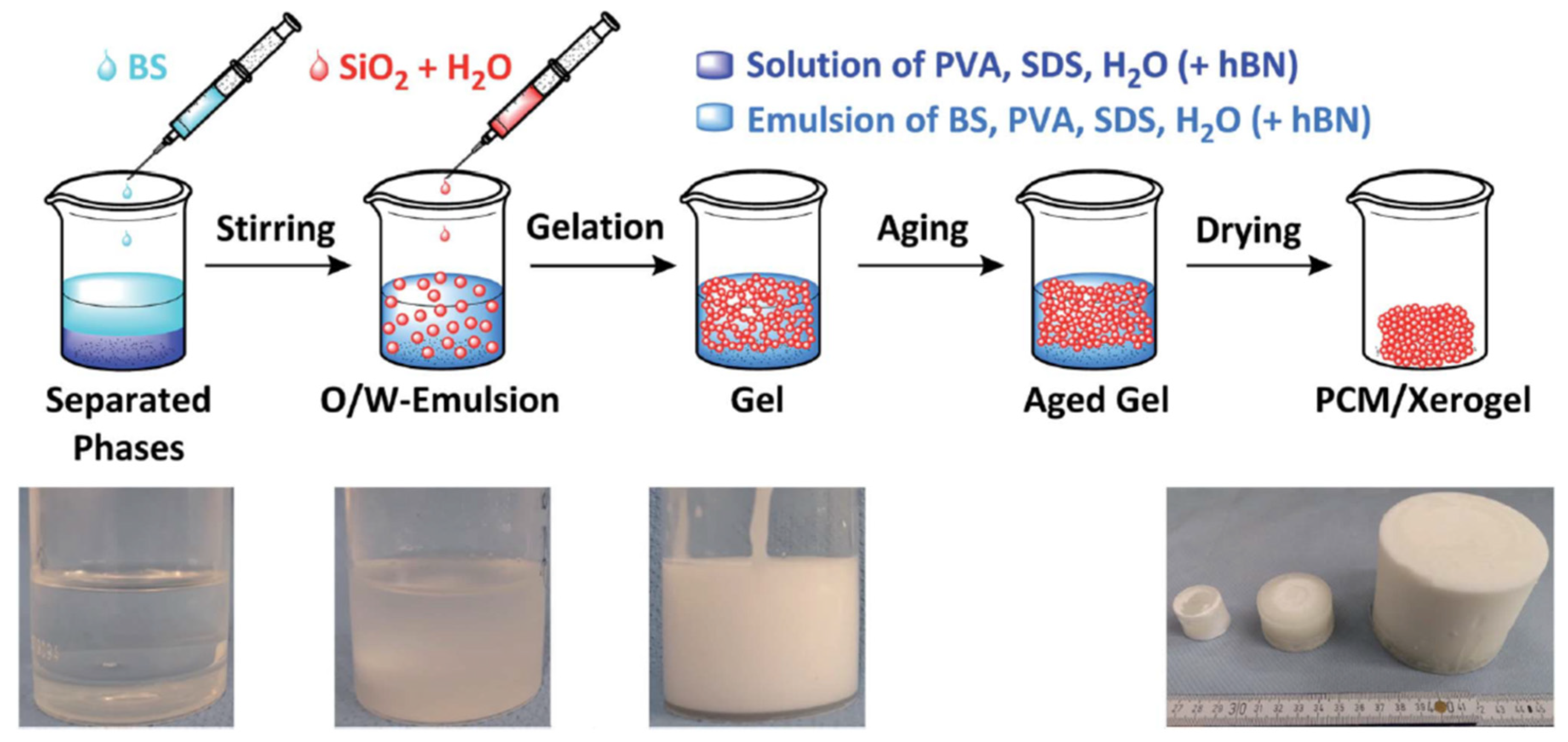
Marske et al. obtained a porous silica monolith containing butyl stearate (BS) using sol-gel synthesis in the presence of SDS and PVA surfactants [
130]. Monolithic and form-stable composites were obtained to a BS loading of 84% wt. (
Figure 9). The 84% BS sample had a compressive strength of 0.7 MPa at 30 °C and higher thermal conductivity than pure BS (0.22 versus 0.12 W m
−1 K
−1). The m.p. of the composite was increased by 2.4 °C with respect to BS, which was explained as arising from the pressure increase in closed pores following the volume change during the phase transition. Heat of fusion values of 80.4 J g
−1 remained unchanged before and after 6000 heating-cooling cycles. The composite monoliths contained both mesopores in the 2–50 nm range and macropores up to 6 μm.
Impregnation of molten fatty acids or derivatives directly into the porous silica matrices is a promising method for obtaining ssPCMs. A ternary eutectic consisting of lauric (LA), capric (CA) and palmitic (PA) acids (CA:LA:PA = 61.9:31.0:7.1) was prepared and used to create composites PCMs using electrospun silica mats as the matrix [
131]. The fibers annealed at 600 °C could adsorb 81% wt. eutectic (
Table 4), with no loss of heat storage efficiency. In a follow-up study, melt impregnation of a quinary fatty acid eutectic was carried out for 10h using electrospun silica nanofibrous mats as the matrix [
132]. The flexible composites exhibited up to 92.2% wt. loading capacity, with shape-stability at 80.2% wt. loading. A ternary eutectic consisting of lauric acid (LA), palmitic acid and paraffin (PAR) with m.p. = 58 °C was prepared in a LA:PA:PAR = 54.25:15.75:30 ratio. The eutectic was then melt impregnated into a disk containing silica nanoparticles obtained from spent lead-acid batteries mixed with high density polyethylene (HDPE) [
133]. The weight fraction of the remaining eutectic after leakage test was proportional with the SiO
2 NPs content, decreasing from 75 to 38% wt. when the SiO
2 fraction decreased from 100% to 0%. Good heat of fusion values were obtained, as well as increased thermal conductivity proportional to the HDPE content [
133].
Melt impregnation of ternary CA:PA: SA = 79.3:14.7:6.0 eutectic into silica nanoparticles was used to prepare composite PCMs for building energy savings applications [
134]. A maximum eutectic loading 75% wt. could be achieved. The composite has good reliability, with only 5% loss in enthalpy after 500 heating–cooling cycles. A commercial fatty ester was impregnated into 16 μm silica gel particles for perishable food storage applications [
135]. While the resulting composite had a PCM loading of 55% wt., the enthalpy only corresponded to 36% of pure fatty ester. Molten caprylic acid was mixed with six types of porous supports (bentonite, natural clay, diatomaceous earth, expanded pearlite, silica gel and activated charcoal) in order to obtain shape-stabilized PCMs [
136]. Expanded pearlite had the highest fatty acid loading at 59% wt., while silica gel incorporated 48% PCM. The heat storage efficiency of the composites was reduced for all samples except expanded pearlite and activated charcoal.
Gas phase transport of lauric acid, capric acid, tetradecanol and dodecanol was investigated using three types of mesoporous SBA-15 with increasing pore diameters from 5.6 nm to 12.5 nm [
109]. Nanoconfined m.p. decrease and non-melting layer formation were noticed.
Fumed silica was impregnated with solution of stearic acid (SA) in chloroform, followed by vacuum distillation [
137]. The SA m.p. was decreased with 0.9 °C with respect to bulk and the amount of acid was computed solely on the basis of heat of fusion (
Table 4). The composite exhibited high thermal reliability and it retained the same heat storage capacity after 600 heating–cooling cycles. Tannic acid templated mesoporous silica having an average pore diameter of 8.4 nm was loaded with stearic acid from ethanol solution at 50%, 60% and 70% wt. [
138]. The 50% loaded sample exhibited only small heat of fusion, indicating that most of the acid is present in the non-melting layer. The 70% wt. loaded sample exhibited two melting events, corresponding to the crystalline nanoconfined and interparticle phases.
Using sacrificial polystyrene nanoparticles, Fan et al. prepared hollow mesoporous microspheres 500 nm in diameter and used them as carrier for stearic acid [
139]. The hollow microspheres were loaded up to 70% wt. with SA. Interestingly, the supercooling exhibited by the fatty acid was reduced, with lower loading yielding lower supercooling. A mesostructured onion-like silica (MOS) material, having large spherical pores up to 50 nm was also used to create stearic acid composite PCMs through ethanol solution impregnation [
140]. 70% wt. was found to be the maximum loading without leakage. A 50% wt. SA loading yielded no heat of fusion, indicating the formation of a non-melting layer. The 70% SA-MOS composite exhibited good reliability after 50 heating-cooling cycles.
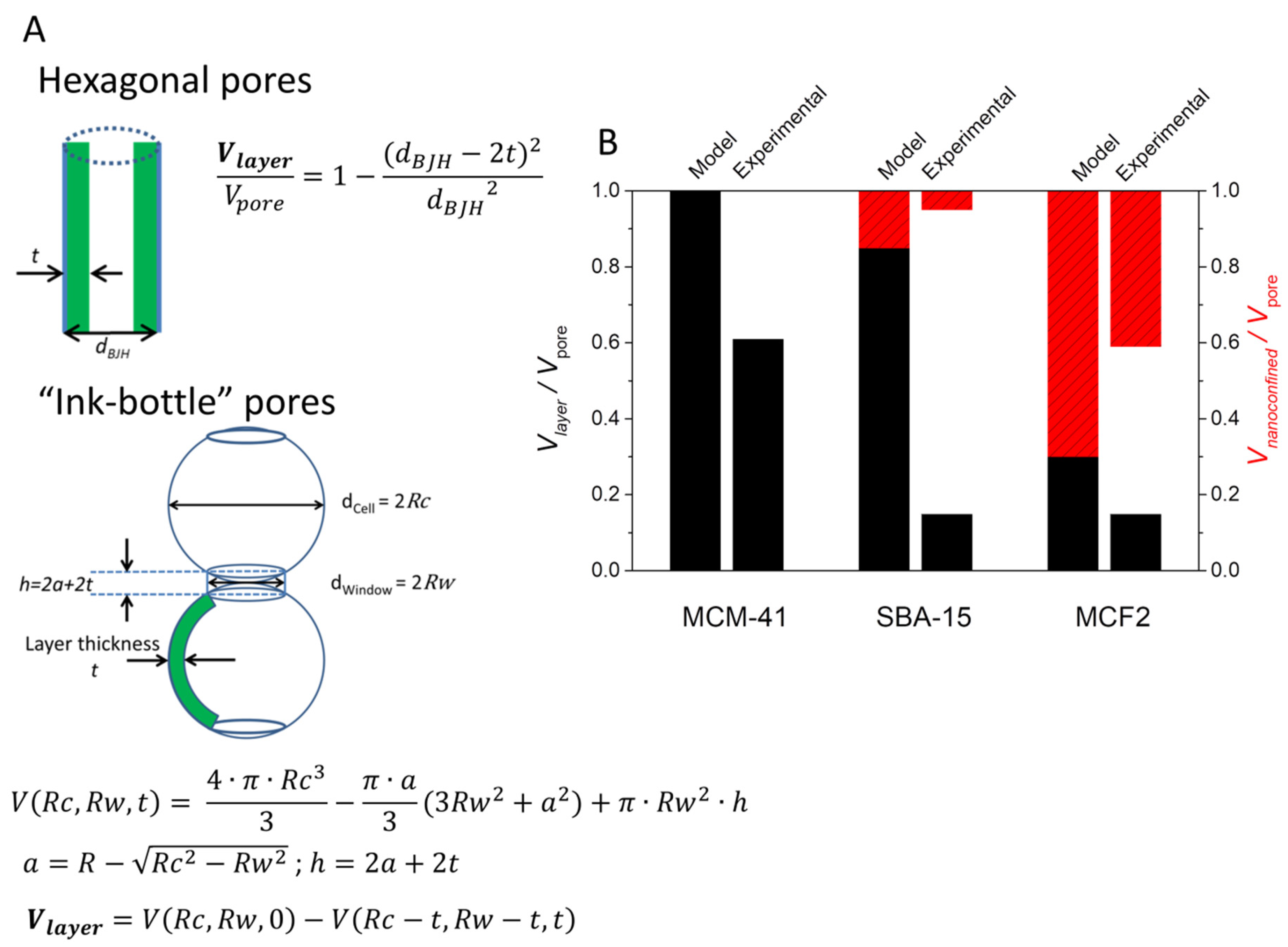
The effect of lauric acid (LA) solution impregnation onto mesoporous silica matrices with varying pore diameters and volumes at different mass fractions was investigated by our group [
67]. MCM-41 and SBA-15 having hexagonally ordered mesopores of 2.7 nm and 6.3 nm and two types of mesocellular foam silica (MCF) with “ink-bottle” pores up to 34.9 nm were used as matrices. LA to silica mass fractions from 1:1 to 6:1 were investigated. Up to 83% wt. LA could be loaded into the 34.9 nm MCF and still retain shape-stability. No heat of fusion was noticed at 1:1 wt. ratio indicating the presence of a significant non-melting layer. All samples containing more than 50% wt. LA showed two melting and two crystallization processes, indicating the presence of both nanoconfined and interparticle phases of the fatty acid. The ratio of the enthalpy of the two processes decreasing with increasing LA fraction, confirming that the first effect is caused by nanoconfinement inside the silica pores (
Figure 10A). Using Equations (2) and (5), the non-melting layer thickness was estimated at 1.8 nm, corresponding to the length of one fatty acid molecule. Based on the thickness, the theoretical non-melting layer volume was computed (
Figure 10A). The difference between the heat of fusion expected based on the LA fraction and the experimentally determined value was attributed to LA molecules present in the non-melting layer (
Figure 10B). The results show that mesoporous silica with average pore diameters less than 20 nm are unsuited for PCM applications, since most of the pore volume will be taken by the non-melting layer. Even for MCF1 with a maximum pore diameter of 34.9 nm, 27% of the total pore volume corresponds to this layer. The impregnation of lauric acid (LA) into mesocellular foam silica (MCF0.5) at 40, 50, and 60% wt. in a different study also showed that no heat of fusion was detected at 40% wt. loading, indicating the formation of the non-melting layer [
141].
A similar study was performed in the case of adipic acid, at three (50, 60, 67%) loading and using MCM-41, SBA-15 and three MCF matrices. [
76] The melting point of the nanoconfined phase was decreased in accordance with Gibbs-Thompson equation. Higher fatty acid loading also influenced the m.p. indicating incomplete mesopore loading, especially for the MCF-type matrices. The adipic acid nanoconfinement was also shown to increase supercooling up to 33 °C for the nanoconfined phase and 15 °C for the interparticle fatty acid phase.
Myristic acid (MA) was loaded through solution impregnation followed by vacuum drying into wrinkled mesoporous silica (WMSN), consisting of radially ordered mesopores which increase in diameter from the core to the shell [
142]. 65% wt. fatty acid composites showed no leakage on a filter paper and good reliability after 50 heating-cooling cycles. The thermal conductivity of the 65% wt. MA composite also increased from 0.27 to 0.37 W m
−1 K
−1 in comparison with the pure fatty acid. The influence of the sebacic acid fraction in composites containing MCM-41 prepared by solution impregnation was tested between 50 and 90% wt. [
143]. Composites with less than 80% fatty acid exhibited shape stabilization. All samples have lower heat of fusion and m.p. values than expected based on the mass fraction of the acid (
Figure 11A). The lost enthalpy is proportional to the MCM-41 content (
Figure 11), while the lowest m.p. was found for the 60% wt. sample. These results indicate that the non-melting layer occupies a significant pore volume and the increasing m.p. can most easily explained as interparticle nanoconfinement due to the lack of two distinct melting events.
Stearic acid, octadecane and octadecanol were used to prepare shape-stabilized PCMs through vacuum melt impregnation using ~200 nm silica nanoparticles [
122]. Shape-stabilized materials with up to 70% wt. PCM could be obtained. No significant melting enthalpy was recovered for PCM loading under 40% wt. for stearic acid and octadecane or 60% for octadecanol. The higher non-melting layer fraction in the case of octadecanol was explained as arising from supramolecular hydrogen bonding between the PCM molecules and the silanol groups present on the nanoparticle surface.
The functionalization of the porous silica surface with various organic groups can affect the properties of the resulting composites, since fatty acids and their derivatives are hydrophobic while the silica surface contains hydrophilic silanol groups. Octadecyl functionalized hierarchically porous monolith containing both ~15 nm mesopores and macropores larger than 1 μm showed the appearance of a nanoconfined stearic acid phase [
107]. Hexamethyldisilazane (HMDS) was employed for creating methyl-functionalized aerogels [
144]. The functionalized aerogels were loaded with palmitic acid or octadecanol through vacuum melt impregnation and compared with pristine aerogels. The methylation treatment increased the pore volume of the aerogel from 3.35 to 4.40 cm
3 g
−1 and average pore diameter from 11.2 to 12.6 nm. The methyl functionalization increased the PCM adsorption capacity with 10–20% wt. and the heat of fusion values with 30–45 J g
−1 in comparison with pristine matrices. The increase could be explained by a synergistic effect of increasing pore volume and diameter coupled with stronger intermolecular interactions between the aerogel pore surface and the hydrophobic PCM molecules.
The effect of functionalization of mesocellular foam silica (MCF) with different organic groups was investigated using stearic acid (SA) as the PCM [
66]. Phenyl and methyl groups were chosen as the hydrophobic aromatic and aliphatic moieties, while propyl amine and propionic acid were employed as the basic and acid hydrophilic functional groups. Molten stearic acid was added to the silica matrices in excess. After cooling, the solids were transferred to filter papers and the excess fatty acid was drained out in order to obtain shape-stabilized composite PCMs. Interestingly, a linear correlation between the pore volume and the SA loading was found. All composites exhibit two melting/crystallization processes corresponding to the nanoconfined and interparticle acid phases, as well as a non-melting interface SA layer. A 2.28 ± 0.29 nm layer thickness was computed based on the m.p. decrease of the nanoconfined phase. The volume occupied by the non-melting layer, the nanoconfined crystalline SA and the empty volume were calculated using Equation (6). These volumes were compared with the experimental values computed from the melting enthalpy. The carboxylic acid functionalization reduced the non-melting layer volume by 25% in comparison with the theoretical value, which could be ascribed to supermolecular interactions between the crystalline nanoconfined acid and the functionalized carboxylic groups (
Figure 12).
The direct sol-gel method can also be used to prepare functionalized silica composites. For example, the hydrolysis and condensation of vinyltriethoxysilane (VTES) in the presence of PVA as surfactant and methyl laurate (ML) was used to prepare composite PCMs for cold energy storage [
145]. While the shape-stability was not reported, composites up to 75% of the heat storage of pristine ML could be prepared. The samples also exhibited increased thermal resistance in comparison with the fatty ester.
The influence of the composites synthesis methods on their thermal properties was studied using lauric acid (LA) and mesocellular foam silica (MCF) [
146]. Ethanol solution impregnation was compared with vacuum melt impregnation, melt filtration, grinding, and pressure treatment. The methods based on solid PCM (grinding, pressure treatment) yielded lower nanoconfined phase enthalpy than the solution or melt syntheses. The pressure treatment was also found to destroy the mesocellular foam pore network. The highest heat of fusion and heat storage efficiency value was obtained in the case of vacuum melt filtration, while the highest nanoconfined phase enthalpy and pore fill percentage were obtained for melt filtration. The pore fill percentage is the percent of the total pore volume of the porous matrix occupied by the nanoconfined crystalline phase and non-melting layer. A portion of the pore volume is empty, since the liquid PCM has lower density than the solid nanoconfined phase.
Improving the thermal conductivity of fatty acid-porous silica PCMs through the addition of carbon based materials was also investigated. For example, 1, 3 and 5% wt. carbon nanotubes (CNT) were added to a 32% wt. capric acid–palmitic acid eutectic adsorbed onto fumed silica [
147]. The CNT do** did not influence the heat storage properties of the composites (other through decreased fatty acid content), but provided an increase of thermal conductivity up to 0.47 W m
−1 K
−1 at 5% wt. CNT versus 0.16 W m
−1 K
−1 for the pure CA-PA eutectic. 1, 3 and 5% wt. graphene nanoplatelets (GNP) were also investigated as an additive for palmitic acid-silica NPs composite PCMs [
148]. The maximum loading without leakage of the PA-SiO
2 material was determined to be 70% wt. Thermal conductivity increases with increasing GNP content, from 0.117 to 0.193 W m
−1 K
−1 for 0% and 5% GNP, respectively. No significant change in enthalpy was noticed after 100 heating-cooling cycles. A graphene oxide-silica mixed aerogel was synthesized and used for both thermal energy storage and light-to-thermal energy conversion [
149]. Octadecanol was melted and impregnated into the aerogel at 75% wt. loading. The composites containing up to 2% GO exhibited lower thermal conductivities than pure octadecanol, but higher than pristine silica aerogels and they could rapidly heat to 50 °C under illumination, proving their light-to-thermal energy conversion. Molten palmitic acid (PA) was impregnated into a hybrid carbon–silica aerogel prepared using 3-aminopropyl triethoxysilane (APTES) as the silica source [
150]. The fatty acid–aerogel composite has similar thermal conductivity values as the pure palmitic acid. An 82.2% wt. acid mass fraction was determined by thermogravimetry. Interestingly, the melting point of the composites with both a carbon aerogel and carbon-silica aerogel is decreased in comparison with bulk, while the enthalpy is similar to the value expected based on mass fraction (
Table 4). These results suggest that the fatty acid is nanoconfined inside the hybrid aerogel without the presence of a non-melting layer.
Obtaining composite PCMs with lower thermal conductivity is also gaining increased attention as a method for thermal insulation and protection. For example, silica aerogels were prepared and loaded with octadecanol (OD) or PEG 2000 as PCMs [
151]. The thermal conductivity of the OD composite was twice as low as that of pure OD, decreasing from 0.25 to 0.12 W m
−1 K
−1 after impregnation. Good heat storage efficiencies were obtained for the composites, with 91% and 96% values for the PEG 2000 and OD-loaded samples, respectively.
Table 4.
Representative porous silica-fatty acids and derivatives nanocomposites.
Table 4.
Representative porous silica-fatty acids and derivatives nanocomposites.
| PCM | Porous Silica Composite | Ref. |
|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Sample/Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
|---|
| Stearic acid | 55.6 | 176.7 | Direct synthesis/TEOS/HCl | 60 | 54.9 | 109.4 | [85] |
| Dodecanoic acid | 44.6 | 169.0 | Gas transport/12.5 nm SBA-15 | - | 22.3 | 65.9 | [109] |
| Tetradecanol | 36.0 | 198.0 | - | 11.4 | 48.4 |
| Decanoic acid | 30.0 | 163.0 | - | 11.1 | 64.3 |
| Dodecanol | 24.0 | 196.0 | - | 0.2 | 69.5 |
| Quinary eutectic | 12.3 | 134.4 | Melt impregnation/electrospun SiO2 fibers | 80.2 | 13.4 | 107.8 | [132] |
| LA:PA:PAR eutectic | 33.1 | 140.6 | Melt impregnation/SiO2 NPs + HDPE | 75 | 31.5 | 104.4 | [133] |
| Lauric acid | 44.4 | 180.8 | Direct synthesis | 65 | 42.5 | 117.2 | [124] |
| Stearic acid | 59.9 | 177.8 | Solution impregnation/fumed silica | 46 | 58.8 | 82.5 | [137] |
| Octadecanol | - | 235 | Vacuum melt/CH3-aerogel | 86 | - | 153.7 | [144] |
| Lauric acid | 42.7 | 166.0 | Hexane solution/MCF | 83 | 34.0/
41.2 | 123.7 | [67] |
| CA: LA:PA = 61.9:31.0:7.1 | 15.0 | 120.2 | Melt impregnation/electrospun SiO2 fibers | 81 | 13.7 | 100.9 | [131] |
| Stearic acid | 65.2 | 239.4 | Solution impregnation/Tannic acid templated SiO2 | 70 | 67.1 | 108.8 | [138] |
| CA:PA: SA = 79.3:14.7:6.0 | 18.5 | 139.3 | Melt impregnation/SiO2 NPs | 75 | 17.2 | 99.4 | [134] |
| CA:MA = 72:28 | 21.7 | 139.2 | Direct synthesis | 40 | 21.15 | 55.6 | [126] |
| Stearic acid | - | 221.8 | Solution impregnation/MOS | 70 | - | 108.0 | [140] |
| Myristic acid | 57.7 | 184.3 | Solution impregnation/WMSN | 65 | 54.7 | 92.0 | [142] |
| Stearic acid | 52.5 | 172.7 | Vacuum melt/SiO2 NP | 70 | 52.1 | 77.6 | [122] |
| Octadecanol | 57.2 | 234.5 | 70 | 56.4 | 47.0 |
| Lauric acid | 42.7 | 176.1 | Vacuum melt/MCF | 84 | 31.5/41.7 | 128.1 | [146] |
| CA-PA (85:15) | 27.5 | 151.5 | Melt/Fumed silica+5% CNT | 30.4 | 25.2 | 41.2 | [147] |
| Stearic acid | 56.6 | 170.3 | Direct synthesis | 76 | 53.8 | 118.3 | [127] |
| Lauric acid | 44.2 | 165.8 | Direct synthesis; TEOS+MTES | | 42.2 | 82.7 | [129] |
| Stearic acid | 68.4 | 213.6 | Melt impregnation/MCF-COOH | 79 | 58.9/68.8 | 128.3 | [66] |
| Methyl laurate | 4.0 | 210.1 | Direct synthesis, VTES/PVA | 71 | 6.7 | 151.3 | [145] |
| Palmitic acid | 62.8 | 209.7 | Solution impregnation; 5% GNP | 70 | 60.6 | 128.4 | [148] |
| Octadecanol | 57.8 | 237.8 | Melt impregnation/2% GO- SiO2 aerogel | 75 | 53 | 129.6 | [149] |
| 1, 8-Cctanediol | 62.4 | 225.1 | Direct synthesis, TEOS/HCl | 70 | 61.3 | 157.7 | [128] |
| Caprylic acid | 12.0 | 139.9 | Melt impregnation/silica gel | 48 | 13.8 | 46.4 | [136] |
| Palmitic acid | 50.0 | 213.1 | Melt impregnation/C-SiO2 aerogel | 82 | 187.7 | 43.4 | [150] |
3.3. Polyethylene Glycol (PEG)-Based PCMs
PEG is considered a suitable material for phase change materials, having large heat of fusion, chemical stability and melting points which depend on the degree of polymerization. As in the case of any PCM based on solid-liquid phase transition, leakage is the main disadvantage which could be overcome by incorporation in various matrices. The confinement effect provided by the silica has an important role in the shape stabilization of the materials. PEG-silica composites represent low-temperature heat storage materials, as PEG has a low melting temperature ranging from 3.2 °C to 68.7 °C [
152].
Yang et al. studied this confinement effect, by preparing PEG-silica composites through a sol-gel method [
153]. Different PEG chains were employed, with M
w of 1500, 4000, 6000, and 10,000 Da. The procedure consisted in dissolving different amounts of PEG in ethanol, followed by the addition of TEOS as silica precursor, in an acidic medium. Shape stabilized composites were obtained at 80 °C when the mass fraction of PEG was between 50–80% in the case of PEG 1500. The sample with 80% content of PEG 1500 exhibited the highest content of crystalline phase and the highest heat of fusion among same PEG chain composites, 7.3 J g
−1, but, much lower in comparison with pure PEG 1500, 148.2 J g
−1. The explanation of this phenomenon is that the lower amount of PEG, the higher its content embedded in amorphous state in the silica, which prevents it from melting. When the content increases, some of the PEG chains have some part out of the silica framework and can undergo melting. The melting point decreased with increasing the amount of PEG, which was associated to nanoconfinement. The FTIR spectra showed that only physical interactions occurred between PEG and silica. The highest crystallinity and heat of fusion was obtained for PEG 10,000, 48.3% crystalline content and 74.5 J g
−1, compared to 167 J g
−1 in the case of free PEG.
Sol-gel synthesis was used to prepare polyethylene glycol M
w 4000 (PEG 4000)-silica composites [
85]. Poor crystallinity for PEG content below 60% wt. was noticed. With increasing the pore size, an increasing in the enthalpy was observed.
The relation between the pore size and the melting temperature of PEG was studied, using porous silica with pore sizes between 10–200 nm [
152]. The correlation is based on the Gibbs-Thompson equation and the authors compared a simple blending method to a solution impregnation one, in order to see the effect of the nanopores on the melting temperature. Two PEG with molar masses of 2000 and 10,000 were blended with or impregnated into disordered porous silica. The crystallinity of PEG decreases with decreasing the pore size, due to nanoconfinement and irregular arrangement of PEG chains on the silica surface. The melting temperature of PEG shifts to lower values with decreasing pore size, in accordance with the Gibbs-Thompson equation, and the enthalpy of fusion also decreases, as the crystallinity decreases (
Figure 13). The supercooling of the composites increases with decreasing pore size, to an extent of almost 6 °C in comparison to the bulk.
A strategy to increase the heat of fusion of PEG/silica composites is to add two types of PEG, with different molecular weights to achieve co-crystallization. Co-crystallization can lead to the increase of the crystalline region because of PEG chains interpenetrations, and therefore an increase in the heat of fusion is obtained. PEG with M
w of 2000 and 10,000 Da was used to prepare shape-stabilized composites through sol-gel synthesis, using TEOS as the silica precursor [
154]. The optimum ratio between PEG 2000 and PEG 10,000 was found to be 3:1, as this composite had the highest heat of fusion, 108.6 J g
−1 (
Table 5). The DSC analyses showed the presence of two melting peaks, associated to the two types of PEG, overlap** to some extent and confirming the co-crystallization.
Guo et al. also used a sol-gel process for the obtaining of PEG/silica composites, starting with Na
2SiO
3 as silica source [
155]. No heat of fusion was noticed at 50% wt. PEG due to the nanoconfinement of amorphous PEG in the silica pores. The enthalpy of fusion increased with increasing the PEG content, getting closer to the theoretical value. The formation of a non-melting PEG layer of constant thickness was used to explain the decreased heat storage efficiency.
Carbon fibers (CF) were introduced to a PEG silica composite obtained through the sol-gel method, using 85% wt. PEG and 1–5% CF wt. content in order to increase its thermal conductivity [
156]. The enthalpy of fusion and melting temperatures of CF/PEG/SiO
2 composites were similar to those of PEG/SiO
2, and lower than that of pure PEG, suggesting that the addition of CF did not exert significant changes on enthalpy or melting temperature. The thermostability of CF containing composites improved when compared to pure PEG. The CF also improved the absorbance of light and its conversion to heat and the thermal conductivities of the composites, proportionally to the CF content. Ca, Mg, and Al metal chloride were used as coagulant in the sol-gel process of obtaining PEG/silica composites, in order to increase the thermal conductivity of the composites without the addition of fillers [
157]. The thermal conductivity of the samples increases with the molar weight of PEG and with the addition of metal ions in the synthesis. The metal cations can form coordination bonds with PEG. The best result was obtained for Ca
2+ ions and PEG 20,000, with a thermal conductivity of 0.41 W m
−1 K
−1. The addition of metal ions, however, leads to lower melting and crystallization enthalpies in comparison to those of silica/PEG composites, because PEG chains movement is hindered by the bonds formed with the metal ions. The materials also exhibit high thermal stability. A method of obtaining PEG/SiO
2 PCMs, starting from silicagel industrial wastes was developed [
158]. As previously noticed, PCMs with 50–70% wt. loading exhibited no leakage, while a small leakage in the case of 80% wt. PEG was noticed. The composites have good thermal stability below 350 °C. The enthalpies of the PEG: SiO
2 PCMs are lower than that of pure PEG, with the highest heat of fusion exhibited by the composite with 80% PEG content, 132.4 J g
−1 compared to 164.6 J g
−1 for pure PEG. For the 50% wt. PEG composite an increase in the thermal conductivity from 0.31 W m
−1 K
−1 to 0.40 W m
−1 K
−1 was noticed.
The presence of a high amount of hydroxyl groups on the surface of silica can affect the thermal capacity of the final composites, because PEG is strongly bound to the surface of the silica through hydrogen bonds, which hinders the crystallization process. Serrano and coworkers studied these interactions by controlling the condensation process, through a second catalysis step, using different quantities of NaOH [
159]. The authors optimized the process for a short gelation time and complete hydrolysis of TEOS, however, the FTIR spectra and DSC analysis confirmed the presence of hydrogen bound PEG. The melting of the PCMs took place at higher temperature than the crystallization process, indicating supercooling. The supercooling was larger for the composites than for pure PEG probably due to the interactions between silica and PEG. The lowest content of non-melting PEG was obtained in the case of the material neutralized with a slight excess of NaOH, because the large content of silanol groups on the silica surface diminish the amount of PEG bound to the OH groups. This composite presented an enthalpy of 113.8 J g
−1, in comparison with 146.7 J g
−1 for pure PEG.
A combined sol-gel method with acrylic acid (AA) in situ polymerization was used for obtaining PEG/SiO
2/AA composites with PEG as PCM and cross-linked silica-AA network as support [
160]. The advantage of this method is that the final material can be molded into any shape, having good shape, thermal stability and thermal reliability. The highest value for the enthalpy of fusion was obtained for the composite with the highest mass fraction of PEG (44.3%), 91.9 J g
−1 versus 171 J g
−1 for free PEG (
Table 5). The measured heat of fusion values are higher than the theoretical ones, which is atypical to these types of composites as they are usually restricted by the nanoconfinement effect. This could be explained by the rearrangement and association of the PEG chains as the crosslinking reaction occurred. The effect of polyacrylic acid on the SiO
2/PEG composites, for different PEG chains was also studied and no increase in enthalpy when compared to the theoretical value was found [
161].
PEG/SiO
2 can be used to control porous asphalt concrete temperature, which can suffer deformation due to high temperatures during summer [
162]. PEG-4000/SiO
2 composites were prepared through a sol-gel process, with the 70% wt. PEG composite having the highest heat of fusion (100 J g
−1). The composite was added into porous asphalt instead of fine aggregates and could reduce the internal temperature of asphalt concrete. A PEG/SiO
2/dye composite was studied for light-to-thermal energy capture, and the results showed that the dye containing composites reduced the degree of supercooling of PEG, and exhibited a melting enthalpy of 167 J g
−1 for 88.5% wt. of PEG in comparison to 212.8 J g
−1 for pure PEG [
163]. This composite showed good light-to-thermal conversion and thermal reliability after 300 phase transition cycles. Wood was impregnated with silica and PEG to obtain a shape stabilized phase change material that can be used as a building material [
164]. The wood was loaded either with only PEG or with PEG/SiO
2. PEG/SiO
2 treated composites exhibited lower enthalpies, but improved the thermal stability, shape stability, and reliability.
Other methods besides the direct sol-gel synthesis can be used for the obtaining silica-PEG composites. PEG impregnations from solution or through vacuum melting have been employed [
165,
166]. Vacuum melting leads to lower heat storage efficiencies due to a higher content of PEG molecules being present in the non-melting layer. In comparison with the sol-gel method, molten impregnation leads to high enthalpy values, being among the highest values obtained in other studies (
Table 5). An impregnation method was also used to prepare composite PCMs [
167], starting from PEG 4000, and a nanoflower-like silica structure. 136.6 J g
−1 heat of fusion values and no leakage were found at 80% wt. PEG. The composite had a melting temperature of 50.8 °C and a crystallization temperature of 41 °C, with the presence of the silica matrix reducing the degree of supercooling.
PEG crystallization behavior can be influenced by different functionalities attached to the silica surface. Dopamine was bound to the silica surface through hydrogen bonding [
168]. PEG (M
w 4000) was then loaded through vacuum impregnation from an ethanolic solution. The polydopamine coating led to the formation of a more crystalline PEG phase, as the heat of fusion of polydopamine composites is higher than the enthalpies of the PEG-silica composites: 73.8 J g
−1 versus 67.2 J g
−1 for 70% wt. PEG. This can be explained by the reduction of PEG-silica surface interaction due to the new hydrogen bonds formed between polydopamine and silica surface, leaving only some imino groups to interact with PEG. The same strategy was adopted for SBA-15 type silica [
169] and the results showed no leaking in the case of the 70% wt. PEG 2000 composite, with good thermal stability. Two endothermic and exothermic peaks were observed in the case of polydopamine composites, which could indicate the presence of a nanoconfined and bulk phase.
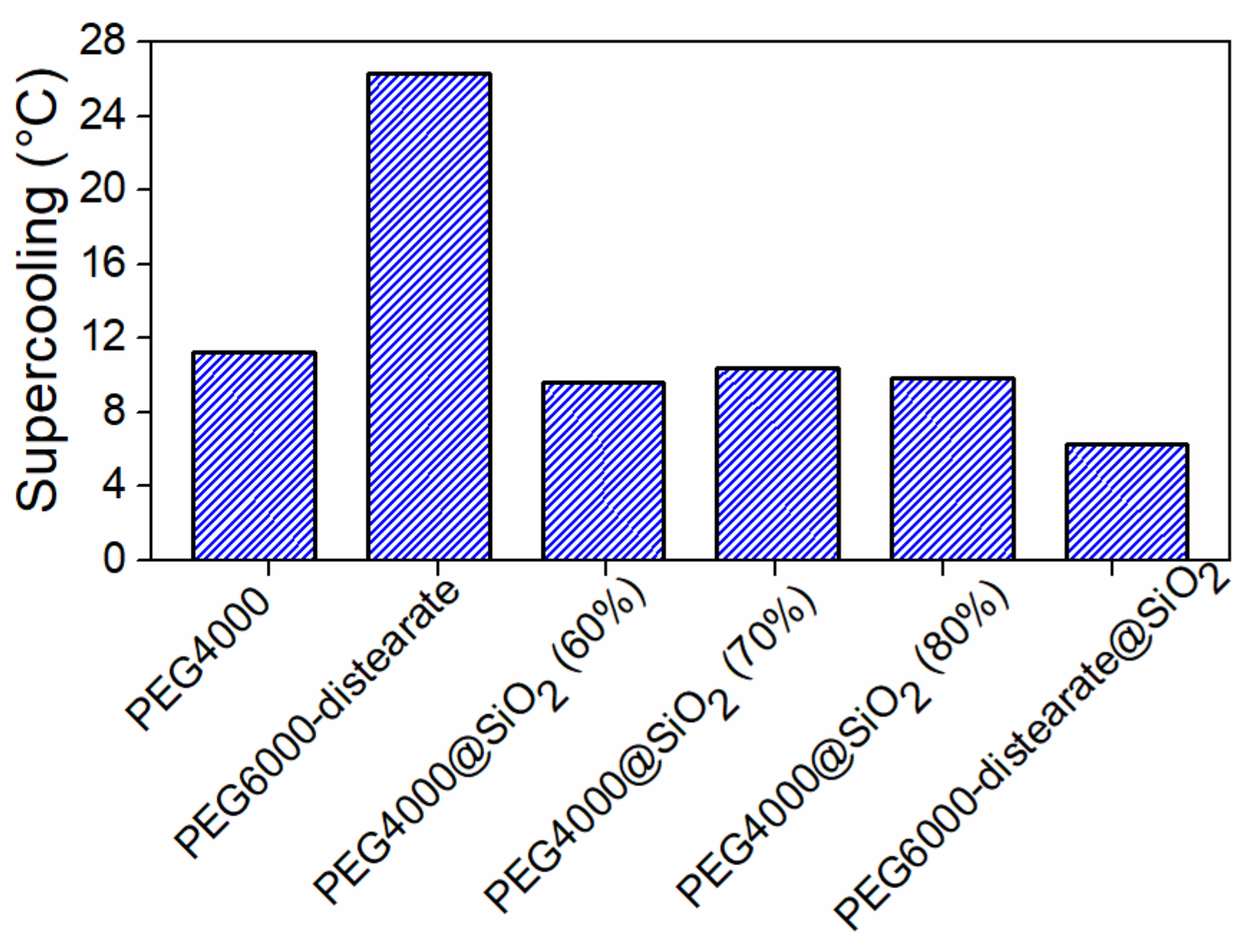
Fatty acid esters of PEG can be used as phase change materials with higher melting points than pure PEG [
170]. 61.6% wt. PEG 6000 distearate was encapsulated into a silica shell through the sol-gel method. The sample showed two phase transitions with temperatures 20 °C and a 52.9 °C and a melting enthalpy of 69.7 J g
−1. The supercooling effect was minimized by the esterification of the end groups of PEG (
Figure 14).
PCMs were also used as additives in cement mortar for buildings external walls in order to reduce the indoor temperature fluctuations [
170,
171]. PEG 600 was impregnated into fumed silica under vacuum. A maximum PEG: silica ratio of 1.6:1 could be loaded with no leakage. These composites were then used in mortar cement mixtures as plaster for several wall specimens, in order to test their properties. The experimental 71.6 J g
−1 heat of fusion value is close to the 72.8 J g
−1 theoretical enthalpy. The presence of the PCM, however, reduces the compressive strength of the mortars, and delays the cement hydration processes.
Li et al. reported obtaining PEG/SiO
2 composites, inspired by mesoporous silica synthesis, in which PEG 6000 is completely encapsulated into a silica shell [
172]. The melting enthalpy of the composite is 164.9 J g
−1, close to that of pure PEG, 178.6 J g
−1 and it corresponds to a mass fraction of 97.3%, calculated from TGA measurements. MCM-41 type mesoporous silica was also used for obtaining shape-stabilized PEG 2000 phase change materials [
173], with PEG loaded through solution impregnation. It was noticed that PEG did not undergo melting even at 70% wt. when impregnated in MCM-41 silica. However, the phase change was noticed starting from 30% wt. when the surface was modified with NH
2 groups. The melting enthalpy of 60% wt. PEG-MCM-41-NH
2 silica composite was 58.8 J g
−1, lower than the theoretical value.
Table 5.
Representative examples of different types of PEG as phase change materials in porous silica composite PCMs.
Table 5.
Representative examples of different types of PEG as phase change materials in porous silica composite PCMs.
| PCM | Porous Silica Composite | Ref. |
|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
|---|
| PEG 600 | 18.5 | 118.2 | solution impregnation | 62 | 19.6 | 71.6 | [171] |
| PEG 2000 | 52.5 | 153.0 | vacuum impregnation | 60 | 50.8 | 58.76 | [173] |
| PEG 4000 | 53.8 | 202.1 | vacuum impregnation | 80 | 50.8 | 136.6 | [167] |
| PEG 4000 | 59.1 | 183.4 | vacuum impregnation | 70 | 57.8 | 121.7 | [166] |
| PEG 4000 | 54.5 | 192.4 | vacuum impregnation | 70 | 53.0 | 73.8 | [168] |
| PEG 6000 | 61.7 | 178.6 | sol-gel | 97.3 | 60.4 | 164.9 | [172] |
| PEG 6000 distearate | 52.9 | 145.1 | sol-gel | 61.6 | 52.9 | 69.7 | [170] |
| PEG 6000 | 59 | 171 | sol-gel | 44.3 | 56.8 | 91.9 | [160] |
| PEG 6000 | 61.4 | 212.8 | sol-gel | 88.5 | 58.4 | 167.0 | [163] |
| PEG 1000 | 35.1 | 146.7 | sol-gel | 84.5 | 35.2 | 113.8 | [159] |
| PEG 1500 | 41.1 | 164.6 | sol-gel | 38.4 | 80.0 | 132.4 | [158] |
| PEG 2000 + PEG 10,000 | 51.7/62.4 | 180.6/
170.9 | sol-gel co-crystallization | 36 | 56.5 | 108.6 | [165] |
The PEG-silica composites show good properties, such as high enthalpy of fusion, thermal stability and reliability and no leakage up to 80% wt., which make them suitable as thermal energy storage materials. They have some drawbacks, such as the large extent of supercooling and strong interaction with the silica surface, but these can be minimized through either PEG chain modification or functionalization of the silica surface.
3.5. Hydrated Salts
The use of hydrated salts as phase change materials is based on their low cost and high heat of fusion. The most known example is the sodium acetate trihydrate, which is found in commercial products. Hydrated salts have large melting enthalpy values, often in excess of 200 J g
−1 with melting points between 0–100 °C (
Table 6). However they also suffer from supercooling and incongruent crystallization, which leads to phase separation between the crystallization water and the salts during heating-cooling cycles. Thus porous silica matrices are also studied with the goal of decreasing supercooling and phase separation.
The addition of silica nanoparticles with diameters between 12 and 50 nm on the thermal properties of Na
2SO
4·10H
2O, Na
2HPO
4·12H
2O, and Na
2S
2O
3·5H
2O was investigated through thermal history and calorimetry [
179]. The addition of up to 7% nanoparticles reduced the supercooling of the hydrated salts crystallization. The MgCl
2·6H
2O-Mg(NO
3)
2·6H
2O system was studied for thermal energy storage [
180]. Compositions between 35 and 50% wt. MgCl
2·6H
2O produced only one endothermic melting peak. 5–20% wt. Fumed silica was then added to the salt mixture, obtaining shape stabilized composites at 15% or higher fractions. The salt mixture containing 41.3% MgCl
2 proved to be unstable in a 100 heating-cooling cycle test, as the water separates from the salts. However, the fumed silica composite shows only a 5% loss of enthalpy after 100 cycles, indicating much better reliability than the hydrated salt mixture. CaCl
2·6H
2O doped with 2% SrCl
2 as a nucleating agent was impregnated into three types of commercial silica nanoparticles [
181]. The maximum salt hydrate loading varied between 70% and 75% wt. depending on the SiO
2 NP size. The heat of fusion of a 75% hydrated salt composite decreased from 148.2 J g
−1 to 138.0 J g
−1 after 500 thermal cycles, indicating high reliability.
Not only inorganic hydrated salts can be used for thermal energy storage. Lan et al. have used
n-alkyl zinc chloride complexes having the formula (
n-C
nH
2n+1NH
3)
2ZnCl
4 as PCMs [
182,
183]. Silica gels with pore sizes varying between 15 and 200 nm were used as matrices for the C
14 complex [
183]. The solid-solid transition of the complex shows the same dependence on pore size as solid-liquid transitions. Both the transition temperature and heat of fusion are decreased with an amount proportional to 1/
d, similar to the Gibbs-Thompson equation.
One of the biggest drawbacks of hydrated salts is their large supercooling and phase separation between the salts and water during use. A comprehensive study using sodium acetate trihydrate as the PCM, carboxymethyl cellulose (CMC) and silica gel as matrices, and Ag nanoparticles as nucleating sites found that the supercooling can be reduced and the heat storage properties can be increased for the optimum composition. This consisted of 85% CMC in the CMC + SiO
2 matrix and 0.5–0.7% wt. Ag NPs [
184]. A silica gel was prepared by the sol-gel method and coated with 10% PVP [
185]. Na
2SO
4·10H
2O-Na
2HPO
4·12H
2O mixture was used as the PCM at 70% wt. loading. The coated matrix exhibited higher stability during cycling, as well as m.p. and heat of fusion decrease in comparison with the hydrated salt fraction, indicating that the stability is caused by the PCM nanoconfinement in the silica matrix. A follow-up study showed that increasing the silica pore size and decreasing the silanol group density yields the highest enthalpy at 70% wt. hydrated salt loading [
186].
The addition of 0.5–4% wt. fumed silica to Na
2HPO
4·12H
2O was studied with the aim of reducing supercooling [
187]. The smallest supercooling degree was found for the 0.5% wt. fumed silica sample, while the thermal conductivity increases with increasing matrix ratio. Hydrophilic fumed silica was added to tetra-
n–butyl ammonium bromide (TBAB) solutions (36–44% wt.) in order to obtain form stable PCMs [
188]. 1.5–3.5% wt. Na
2HPO
4·12H
2O was added as a nucleating agent, while the fumed silica ratio was varied between 20–35% wt. The addition of 2.5% Na
2HPO
4·12H
2O yielded composites with the lowest supercooling degree while 30% fumed silica was sufficient to obtain form stable materials with no leakage. The total heat storage capacity of the composite decreased from 134.0 to 111.6 J g
−1 after 100 heating-cooling cycles. A similar amount of fumed silica (30%) was also needed to obtain shape-stabilized PCMs using the Na
2SO
4·10H
2O-Na
2HPO
4·12H
2O eutectic and Na
2SiO
3·9H
2O as nucleating agent [
189].
A non-eutectic mixture of urea and sodium acetate trihydrate was melted and mixed with fumed silica as the matrix, 1.5% wt. Na
2HPO
4·12H
2O as nucleating agent and 2% wt. sucrose as thickener [
190]. 30% wt. silica was sufficient to prevent leakage and decreased supercooling to 1.1 °C. In addition, the sample had good reliability after 200 heating-cooling cycles, with a loss of only 2% of its enthalpy.
The decrease of the available pore volume caused by the presence of the non-melting layer was used to quantify the nanoconfined enthalpy of hydrated salts PCMs adsorbed into SBA-15 [
191]. The non-melting layer was approximated using the fractal dimension model (see
Section 2.4).
Table 6.
Representative examples of hydrated salts as phase change materials in porous silica composite PCMs.
Table 6.
Representative examples of hydrated salts as phase change materials in porous silica composite PCMs.
| PCM | Porous Silica Composite | Ref. |
|---|
| Sample | m.p. (°C) | ΔHf
(J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf
(J g−1) | |
|---|
| Na2SO4·10H2O-Na2HPO4·12H2O | 36.7 | 226.9 | Melt impregnation/sol-gel SiO2 + PVP | 70 | 30.1 | 106.2 | [185] |
| Melt impregnation/sol-gel SiO2 | 70 | 28.5 | 67.5 | [186] |
| MgCl2.6H2O:Mg(NO3)2·6H2O (41.3:58.7) | 58.8 | 118.5 | Melt impregnation/fumed silica | 85 | 54.3 | 88.1 | [180] |
| Na2SO4·10H2O-Na2HPO4·12H2O | - | 221.4 | - | 70 | - | 64.1 | [191] |
| TBAB:H2O (4:6) | 11.8 | 211.9 | Melt impregnation/hydrophilic fumed silica | 70 | 8.3 | 134.0 | [188] |
| CaCl2·6H2O | 29.4 | 199.9 | Melt impregnation/SiO2 NPs | 75 | 25.1 | 148.2 | [181] |
3.6. Molten Salts
Molten salts are typically employed for high temperature applications as PCMs. Most molten salts are corrosive and can react with the silica matrix at elevated temperatures, thus a careful selection of the salt nature, silica matrix and operating temperature must be carried out.
A 1:1 wt. sample consisting of sodium sulfate and silica was prepared by sol-gel synthesis starting from sodium silicate [
192]. The pore diameters are around 20 nm in the silica framework. The composite exhibits gradual loss of enthalpy upon repeated heating and cooling, with 15% of its heat storage capacity lost after 100 cycles. The direct sol-gel method was also used to prepare a 60% wt. NaNO
3/SiO
2 composite starting from sodium silicate [
193]. The composite maintains its shape at elevated temperatures and it has thermal stability up to 500 °C. Na
2SO
4/SiO
2 particles were embedded into SiO
2-Al
2O
3 aerogels up to 61% wt. loading [
194]. The unloaded aerogel had a pore volume of 4.28 cm
3 g
−1 while the PCM sample still retained 0.57 cm
3 g
−1 total pore volume even after treatment at 1000 °C. A total enthalpy of 125 J g
−1 with a melting point of 871.2 °C was obtained for the composite containing 61% wt. Na
2SO
4/SiO
2 particles.
The sol-gel method was also employed to obtain Li salts-silica composites with salt loading up to 60% wt. [
195]. While Li
2CO
3 and CH
3COOLi·H
2O did not yield shape-stabilized PCMs due to reactions with the silica framework, LiCl and LiNO
3 could be used for heat storage applications. In particular, the LiNO
3-SiO
2 composite prepared using 60% wt. salt retained 236.3 J g
−1 after 50 heating-cooling cycles.
The addition of 10% wt. SiO
2 or SiC nanoparticles to a mixture of Na
2CO
3-K
2CO
3 eutectic containing 45% wt. MgO was investigated [
196]. The sintered samples containing the SiO
2 nanoparticles exhibited similar m.p. and thermal conductivity values as the starting material, while the heat of fusion was reduced due to the lower eutectic mass fraction (
Table 7).
KCC-1 mesoporous silica with radially ordered pores which increase in diameter from the center of the particles to the exterior has been used to create PEG, LiNO
3 and Na
2SO
4 composite PCMs [
166]. 70% wt. loadings resulted in shape-stabilized materials, which exhibited little heat loss after 20 heating–cooling cycles and high heat storage potential (
Table 7).
A study of NaCl-CaCl
2 eutectic impregnation into mesoporous silica with 6.3 and 8.1 nm mesopore diameters was performed by our group [
197]. The initial eutectic salt fraction was varied from 70–95% wt. in the final composite. No melting/crystallization processes were noticed for salt loadings lower than 90% wt. This phenomenon arises from the reaction of calcium chloride with the silica matrix, yielding calcium silicate species. An initial NaCl-CaCl
2 loading higher than 80% wt. is required for the complete formation of the calcium silicate matrix. The resulting composites have specific heat capacity of 1.0–1.1 J g
−1 K
−1, thermal stability up to 700 °C, and shape-stability above their melting point, indicating that the mesoporous silica can be used as a reactive matrix for obtaining shape-stabilized PCMs containing molten salts.
A 1:1 mol. molten NaNO
3-KNO
3 eutectic was adsorbed into five types of mesoporous silica nanoparticles at 90% wt. [
74]. The MSN matrices consisted of hexagonal 2.7 nm pore diameter MCM-41 and 6.3 nm diameter SBA-15, as well as FDU-12 with a cubic arrangement of “ink-bottle” 3.7/9.0 nm diameter mesopores and two types of mesocellular foam silica (MCF) with 9.7/22.4 nm and 13.2/29.8 nm diameter disordered pores. With the exception of MCM-41, all other MSN yielded shape-stabilized materials. Interestingly, two melting and two crystallization processes were evidenced, corresponding to the mesopore nanoconfined and interparticle salt phases (
Figure 15A,B). The heat of fusion decrease with respect to the theoretical value based on the eutectic mass fraction indicates the presence of a non-melting interface layer. Using the m.p. depression and Gibbs–Thompson equation, the thickness of the non-melting layer was computed at 1.9 nm (
Figure 15C). Using the model presented in
Figure 5, the pore volume fractions occupied by the non-melting layer, the crystalline nanoconfined salt phase and the empty pore volumes were calculated and compared with the results determined from experimental measurements (
Figure 15D). These results show a direct increase in nanoconfined volume and decrease of the non-melting layer with increasing pore diameter, as well as a corresponding reduction of the empty pore volume, suggesting that mesoporous silica with larger pore diameters are the best matrices for molten salt PCMs.
Melt impregnation was also used to load a ternary NaCl-NaBr-Na
2MoO
4 salt eutectic into various mesoporous silica matrices at 80% wt. mass fractions [
198]. This molten salt eutectic exhibits both a solid-liquid transition at 522 °C and a solid-solid phase change at 454 °C which could be used for high temperature heat storage. High temperature optical microscopy confirmed that all composites retain their shape at temperatures higher than their melting points. The m.p. of each composite is decreased with 5–10 °C with respect to the pure eutectic, signifying nanoconfinement effects in the interparticle space. Similar decreases are noticed for the solid-solid transition as well. Using the Gibbs-Thompson equation, the diameter of the interparticle salt phase was computed at 60–150 nm, higher than the 2.7–29.8 nm pore size. All composite samples have high thermal stability up to 700 °C. The sample obtained using MCM-41 mesoporous silica matrix with the smallest pore diameter (2.7 nm) exhibited high total heat of fusion (168.9 ± 8.7 J g
−1) which remain unchanged after 50 heating-cooling cycles. The morphology of the mesoporous silica matrices varies from agglomerated rods for SBA-15 to spheres for mesocellular type silica (
Figure 16). The samples containing the ternary molten salt mixture were shown to have an even distribution of the salt species and the silica matrices using electron microscopy analyses (
Figure 16).
A comparative study between direct sol-gel synthesis and melt impregnation has been carried out using Na
2SO
4·10H
2O as the starting salt [
199]. The sodium sulfate fraction was varied between 20% and 60% wt. in the sol-gel syntheses. The heat of fusion values agree with the initial salt ratio. Vacuum melt impregnation into silica gels yield similar enthalpies and melting points, suggesting that the silica surface silanol groups do not influence the heat storage process.
Table 7.
Representative molten salt-porous silica composites for thermal energy storage.
Table 7.
Representative molten salt-porous silica composites for thermal energy storage.
| PCM | Porous Silica Composite | Ref. |
|---|
| Sample | m.p. (°C) | ΔHf (J g−1) | Synthesis | %PCM (wt.) | m.p. (°C) | ΔHf (J g−1) | |
|---|
| Na2SO4 | 888.7 | 167.1 | Direct sol-gel synthesis | 50 | 886.0 | 82.3 | [192] |
| NaNO3 | 308.0 | 189.0 | Direct sol-gel synthesis | 60 | 302.0 | 108.0 | [193] |
| LiNO3 | 253.8 | 369.9 | Melt impregnation/KCC-1 | 70 | 251.3 | 292.2 | [166] |
| Na2CO3-K2CO3/MgO | 702.9 | 81.4 | Sintering/SiO2 NPs | 90 | 703.6 | 76.2 | [196] |
| NaCl-CaCl2 (1:1 mol) | 499.5 | 208.2 | Reactive melting/8.1 nm MSN | 95 | 499.1 | 60.8 | [197] |
| Na2SO4 | 886.7 | 167.1 | Direct sol-gel synthesis | 60 | 886.9 | 100.8 | [199] |
| LiNO3 | - | - | Direct sol-gel synthesis | 60 | 232.8 | 236.3 | [195] |
| NaNO3:KNO3 (1:1 mol) | 221.8 | 96.9 | Melt impregnation/MCF | 90 | 201.0
221.2 | 78.7 | [74] |
| Na(Cl, Br, MoO4) | 454.5
522.1 | 78.9
137.3 | Melt impregnation/MCM-41 | 80 | 450.4
514.4 | 52.8
111.2 | [198] |


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















