2.1. Nitrite Reductases (NiR)
NrfHA menaquinol: nitrite oxidoreductase complex catalyzes the six-electron reduction of nitrite to ammonia in a reaction that involves eight protons. It houses a total of 28 heme groups in the biological unit and, as such, represents a challenge for every experimental approach. Twenty-two heme groups have 6cLS and six have 5cHS configuration (
Figure 3a); two of the HS hemes are membrane-integrated and the other four represent catalytic sites that carry unusual Lys-coordination. The HS and LS hemes can be easily distinguished in the RR spectra of NrfHA, with redox-sensitive
ν4 and redox/spin-sensitive
ν3 modes of the HS species at 1366 and 1493 cm
−1, and of the LS at 1373 and 1501 cm
−1, respectively (
Figure 3b), allowing for independent monitoring of the processes that involve these two populations. Binding of nitrite to the catalytic HS hemes and the resulting spin configuration of the initial enzyme/substrate complex have profound consequences for the reaction mechanism of nitrite reduction, indicating whether the N-O bond cleavage follows the homolytic or heterolytic route, and the subsequent steps of the catalytic cycle. RR data have provided the first experimental evidence that nitrite binding to NrfHA-active site HS hemes causes a spin conversion from HS to LS configuration, which implies that the heterolytic cleavage of the N-O bond is favored in the first step of the catalytic reaction [
23].
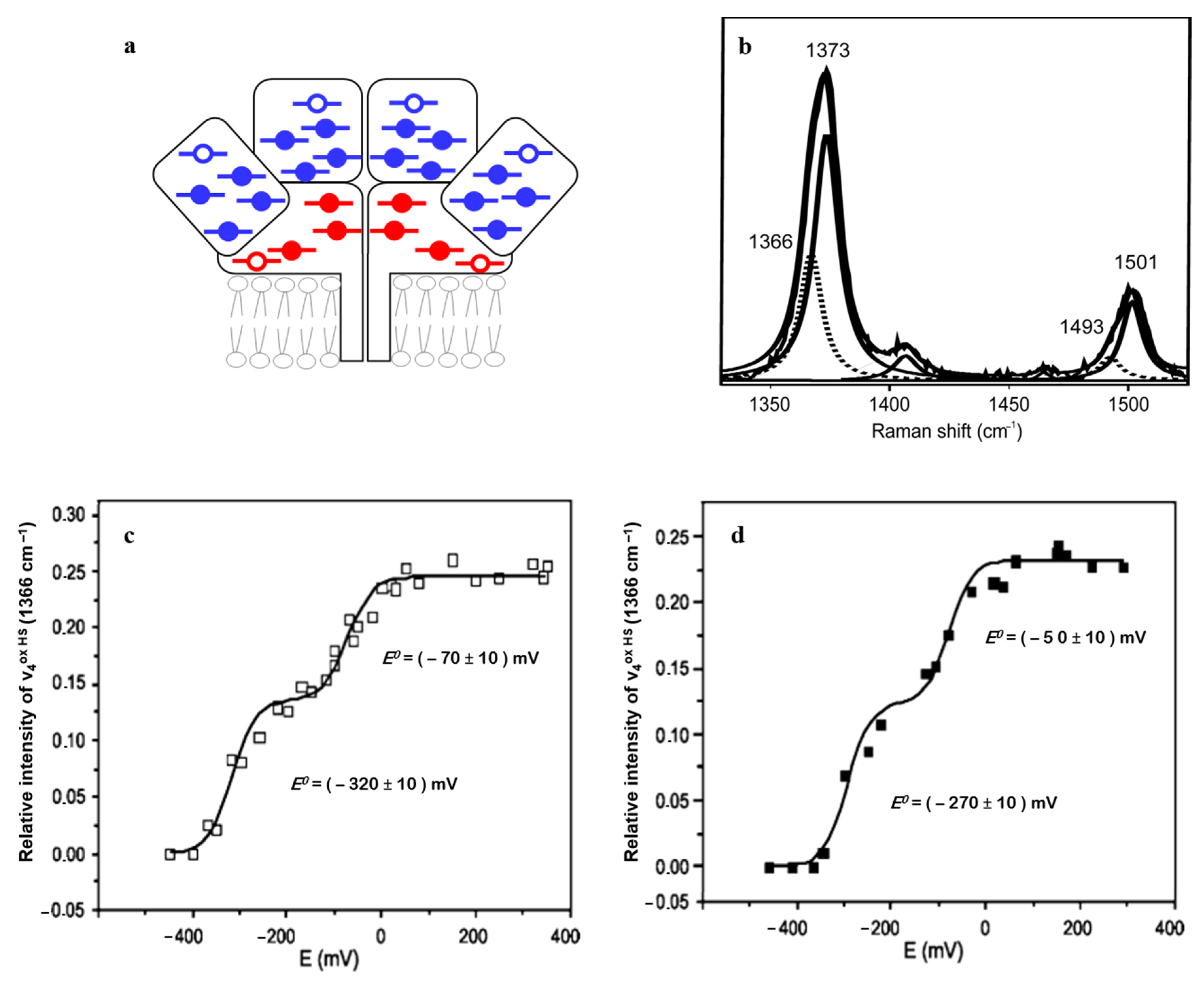
SERR spectro-electrochemistry furthermore helped disentangle the ET pathway in NrfHA. Potentiometric titrations of NrfHA immobilized on biocompatible Ag electrodes was followed by the analysis of the ν
4 band of the HS hemes only (1366 cm
−1) in the presence/absence of 2-
n-heptyl-4-hydroxyquinoline
N-oxide (HQNO). The inhibitor selectively binds in the proximity of the membrane-integrated HS heme subpopulation (red empty symbols,
Figure 3a). Out of the two redox transitions, observed at
E0 = −270 ± 10 mV vs. NHE and at
E0 = −50 ± 10 mV vs. NHE, only the former was modulated in the presence of HQNO (
Figure 3c,d). The
E0 which remained unaltered in the presence of the inhibitor was therefore assigned to the catalytic NrfA HS hemes. These SERR-based insights provided the first evidence for the downhill biological electron flow in the integral NrfHA [
6].
Cytochrome cd1 nitrite reductases (
cd1NiRs) catalyze the reduction of nitrite to NO as a part of bacterial dissimilatory denitrification pathway. They are homodimer proteins containing a
d1-type heme in the active site and a
c-type heme as ET center in each subunit [
24,
25]. Owing to their distinctive electronic absorption properties, the hemes
c and
d1 can be selectively probed by RR spectroscopy employing 413 (or 514 nm) and 457 nm laser excitation, respectively [
26,
27,
28]. Upon binding of the reaction product, NO, the Soret band of heme
d1 (ca. 460 nm) is blue shifted, allowing for both hemes to be probed with 413 nm excitation [
28]. RR spectra of
cd1NiR-NO adducts revealed that in addition to the heme
c, two
d1-heme spin configurations co-exist: a 6cLS-NO species and a 5cHS-NO state, in which the proximal His residue is detached from the heme [
28]. The presence of the 5c adduct was confirmed by a 520 cm
−1 mode whose frequency falls in the range of Fe-NO stretching modes characteristic for 5c-NO adducts of
c-type cytochromes [
29,
30,
31]. Conversely, the stretching coordinates of the 6cLS-NO
d1 heme have been observed at unusually high frequencies (585 cm
−1 in
Pseudomonas aeruginosa cd1NiR) in comparison to other heme–NO complexes [
29,
30], which is thought to be related with the electronic properties and highly ruffled structure of heme
d1 [
27].
2.2. Heme-Containing Respiratory Chain and Analogous Complexes
Complex III (ubiquinol: cyt
c oxidoreductase or
bc1 complex) catalyzes the transfer of two electrons from ubiquinol to two cyt
c molecules.
bc1 complexes are formed by a minimum of three subunits, one contains a
c1-type heme (or structurally and functionally analogous cytochrome
f in plants, cyanobacteria, and green algae that house cytochrome
b6f complex), one holds a Rieske type [2Fe-2S] cluster, and the third one contains two LS
b-type hemes, designated
bL and
bH [
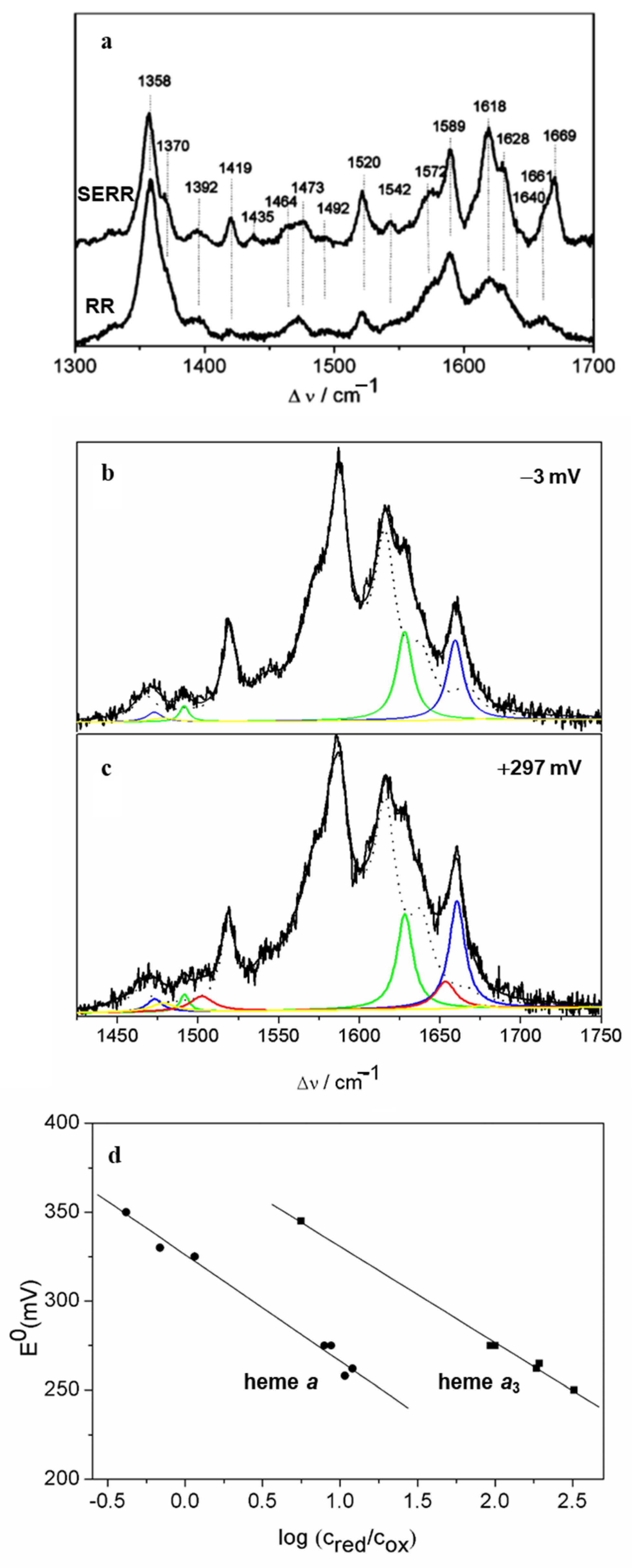
10]. RR spectroscopy has been employed in the initial characterization of heme groups of the complex at different stages of reduction. Namely, due to the differences in midpoint redox potentials of the heme
c1, low-potential heme
b (
bL) and high-potential heme b (
bH), it is possible to selectively reduce the higher-potential sites (heme
c1 and
bH). A selective resonance enhancement using multiple excitation lines and sequential stoichiometric reduction of the complex allowed for spectral distinction between the
c and
b-type hemes, providing insights into the differences in peripheral heme–protein interactions, and in particular the conformation of vinyl substituents of the pyrrole rings [
10].
RR spectra of cyt
c1- and cyt
f-containing subunits of
bc1 and
b6f complexes employing Q-band (550 nm) excitation effectively probe the respective local heme environments of these sites, indicating remarkably similar macrocycle geometry of the two hemes [
32]. Cyt
b6f participates in the oxygenic photosynthesis as a redox link between the two reaction center complexes. RR spectroscopy initially helped identify the chromophoric groups in
b6f complexes isolated from different species, revealing a presence of chlorophyll
a,
β-carotene, and an additional 5cHS
c1-type heme [
33]. RR spectra of oxidized, native, ascorbate-and dithionite-reduced forms of spinach cyt
b6f, obtained using 441, 413, and 406 nm lasers, reveal RR contributions of chlorophyll
a,
β carotene, the 5cHS
c-type heme of cytochrome
f, and the
b-type hemes of cytochrome
b6 of the complex [
34]. RR bands arising from the pigments, found in 1520–1575 cm
−1 range, are particularly intense in the spectra obtained with 441 nm [
34]. Different conformations of the two
b-type hemes, a strongly distorted vs. largely planar geometry, were observed and correlated to the differences in the redox potentials of the two hemes in cyt
b6f, which appears to be a common feature for the
bc1 and
b6f complexes. A functional analogue to
bc1 complex carrying two
b-type hemes and one
a-type heme was found in an archaeon. RR spectroscopy helped characterize the heme groups in this enzyme that show
bc1 complex activity [
35].
IR difference spectroscopic studies of the
bc1 complex revealed redox-induced structural changes, while additional information obtained on site-directed mutants and site-directed labeling of cofactors ensured complete assignments of the observed vibrational modes [
36]. More recently, ATR-IR spectroscopy was employed to study redox changes in thin layers of bovine
bc1 complex that have been deposited on the surface of a silicon microprism. Thereby, redox-sensitive IR absorption bands in the potential-induced difference spectra of
c1,
bH, and
bL hemes, ubiquinone, and surrounding amino acid residues were identified upon selective reduction. A similar approach was used to probe the [2Fe–2S] cluster of the complex. The pH-dependent IR features in the reduced minus oxidized difference spectra revealed specific signals that were attributed to an imidazolate-to-imidazole transition, providing the first experimental evidence that a cluster coordinating His is the most likely candidate for redox-linked protonation site [
37,
38].
Cytochrome c peroxidase, C
cP: Soluble, periplasmic, dihemic C
cPs catalyze the reduction of hydrogen peroxide to water by two electrons delivered from small redox proteins in some bacteria. These C
cPs contain two
c-type hemes, one which is in analogy to Complex III, high-potential, H-heme (
E0 = 330 to 450 mV vs. NHE), and the other low-potential, L-heme (
E0 = −330 to −250 mV vs. NHE) in the active site. The resting state of a dihemic C
cP is typically a catalytically inactive diferric form, with a His- and Met-coordinated H-heme that participates in ET to the active site, and catalytic bis-His coordinated L-heme that requires activation. It is achieved by reduction of the H-heme, which induces Ca
2+-dependent spin and coordination change of ferric L-heme to a 5cHS state upon distal Fe
L-N (His) bond disruption. RR spectroscopy helped characterize these conformational changes on the level of the two hemes that occur in some C
cPs in parallel with those rare C
cPs that do not require activation [
7,
8,
9]. More recently, RR experiments revealed that some C
cPs do not follow either of the abovementioned scenarios, as no significant amount of 5cHS ferric heme population could be detected either in diferric or semi-reduced states. This was rationalized in terms of a novel, more subtle activation mechanism that likely involves formation of a 6c hydroxo complex, which could react with hydrogen peroxide to create the ferric hydroperoxo complex [
7,
8,
9].
Oxygen reductase—Complex IV (cyt
c: oxygen oxidoreductase, C
cO, or heme copper oxygen reductase, or heme copper oxidase, HCO) catalyzes the reduction of molecular oxygen to water by utilizing four electrons and four protons. This is one of the most fundamental reactions in living organisms. HCOs pump protons from the N to the P side of the membrane, contributing to the generation of transmembrane electrochemical potential, which drives the ATP synthesis [
39]. The catalytic reaction occurs at a binuclear center formed by a HS heme (e.g., heme
a3 in mitochondrial enzyme, here designated as C
cO) and a copper atom (Cu
B). The ET to the binuclear center is mediated by a LS heme group in the catalytic subunit (heme
a in C
cO) and a copper center in the non-catalytic subunit, which is composed of two copper atoms (Cu
A) that hold one redox equivalent (
Figure 4a middle). The number of non-catalytic subunits and their cofactors vary in HCOs. Bacteria and archaea have complexes that are simpler than the mammalian C
cO, and if required, the expression of the appropriate HCO (e.g.,
aa3-,
cbb3-, or
ba3- type) can be fine-tuned as a function of oxygen pressure levels in the environment. RR spectroscopy played a fundamental role in identification and description of the cofactors in these enzymes, such as (i) the type and spin, oxidation, and coordination state of the heme groups in structurally diverse HCOs of different origin [
40,
41,
42,
43,
44] and (ii) interactions, conformations, and the dynamics of small ligand (CO, CN
−, N
3, NO) binding [
45,
46,
47,
48,
49,
50,
51].
The low-frequency region of RR spectra has provided unprecedented details on (i) mechanistic properties of HCOs, including detection and identification of the short living catalytic intermediates formed upon oxygen binding to the catalytic HS heme (i.e.,
a3,
b3, o3) and (ii) molecular environment and conformation of the catalytic site, based on correlation of Fe-CO and Fe-C-O stretching mode frequencies of HS heme-CO adduct [
52,
53]. Moreover, protonation events during the catalytic reaction and in particular the role of the two HS heme propionates and the highly conserved amino acid residues found in their vicinity, have been elucidated by RR spectroscopy using structurally different HCOs [
50,
54], isotopic substitution, wavelength selective resonance enhancement, and mutagenesis studies [
55]. A presence of
a-type hemes in some HCOs offers further advantages as the effect of mutations and other molecular perturbations can be monitored via porphyrin formyl stretching C=O modes, which are well resolved in RR spectra of ferrous HS and LS hemes at 1661 and 1628 cm
−1, respectively. In this manner, the frequencies of
a (C=O) and
a3 (C=O) stretching modes reveal the respective heme environments [
56]. They highlight the importance of H bonding interactions stabilizing the heme
a and hydrophobic environment surrounding the heme
a3 in an
aa3-type HCO and the role of the specific amino acids that participate and ensure a proper environment and H network [
54].
Perhaps the most significant contribution of RR spectroscopy of heme proteins can be attributed to the understanding of the complex catalytic reaction of HCOs, as it provided identification and structural characterization of the transient intermediate species and kinetics of their formation. Reduced HCOs bind molecular oxygen and reduce it via formation of short living A, P, F, and H intermediates [
57], which were initially only tentatively described by electronic absorption spectroscopy. RR experiments performed independently by the groups of Kitagawa, Rousseau, and Babcock led to a consistent mechanistic model of HCOs, employing isotopic labeling, mutagenesis studies, a number of structurally different HCOs, and individually developed TR RR experimental approaches [
39,
57,
58,
59,
60,
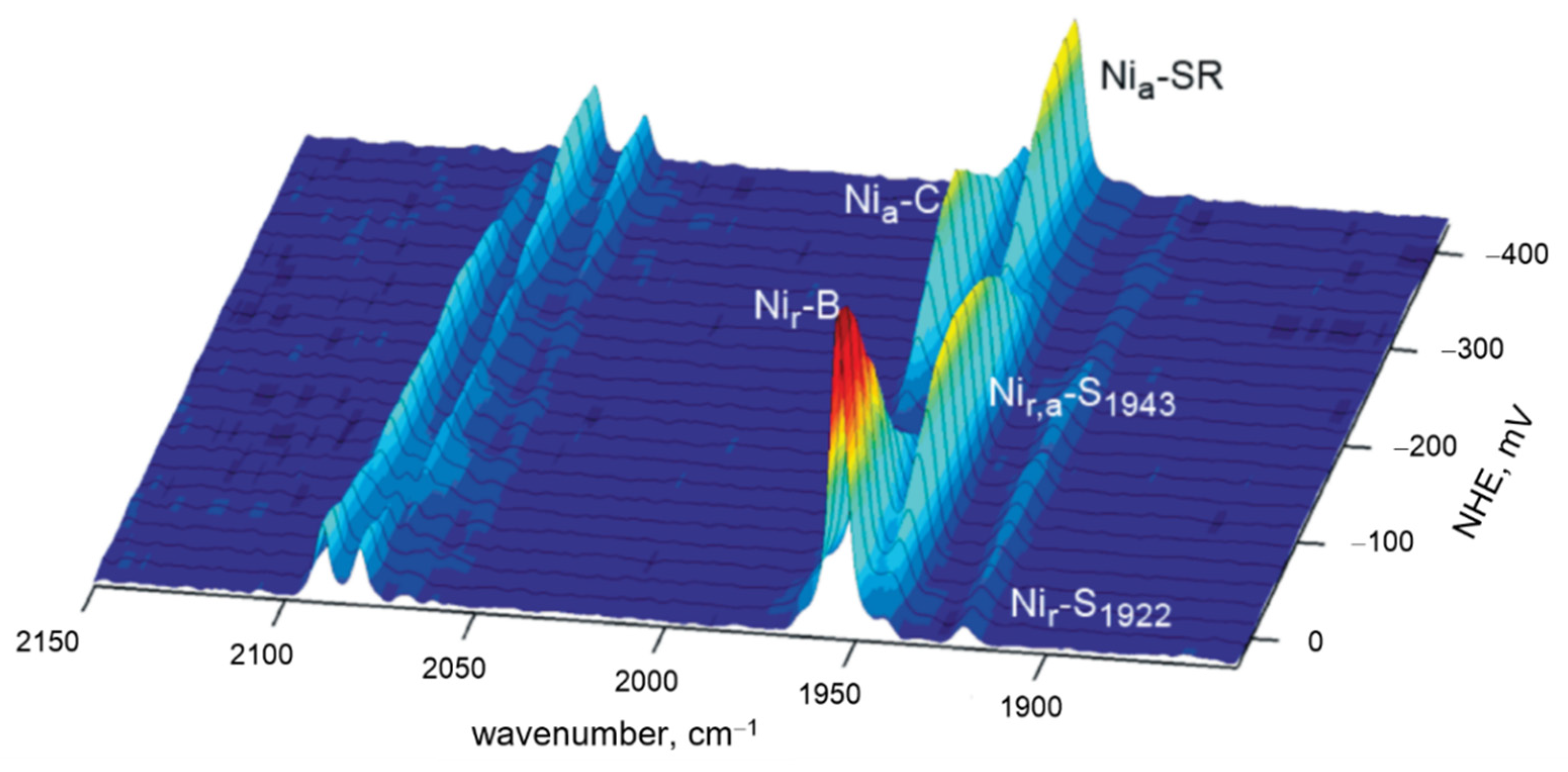
61]. In particular, iron–oxygen stretching frequencies obtained by TR RR
16O
2–
18O
2 difference spectra, measured in H
2O and D
2O media, helped identify proton-coupled ET reactions and establish structural fingerprints of each catalytic intermediate (
Figure 4b). These include the first formed compound A, assigned to ferric-superoxide species with iron–oxygen stretching frequency at 568 cm
−1, compound P (iron–oxygen stretching frequency at 804 cm
−1, electronic transition at 607 nm), and compound F (iron–oxygen stretching frequency at 786 cm
−1, electronic transition at 580 nm). The P and F are both attributed to oxoferryl, Fe
4+ = O, species with subtle differences in the proximal His ligand of
a3 and one of its propionates, and/or the presence of a nearby amino acid (Tyr) radical [
57,
62]. The intermediate F decays into hydroxyl H species, with Fe-OH stretching frequency of 450 cm
−1,
Figure 4b.
IR difference spectroscopy recorded under steady state conditions and in the TR mode identified protonation/deprotonation and re-protonation events during the catalytic cycle of
aa3-type HCO. The data in particular point out the role of a proton shuttle of glutamic acid found at ~11 Å from the active site, which changes its protonation state a number of times during the catalytic cycle. The role of the Tyr located in the proximity of the binuclear center in the splitting of the O-O bond during the catalysis was also highlighted by IR spectroscopy [
63].
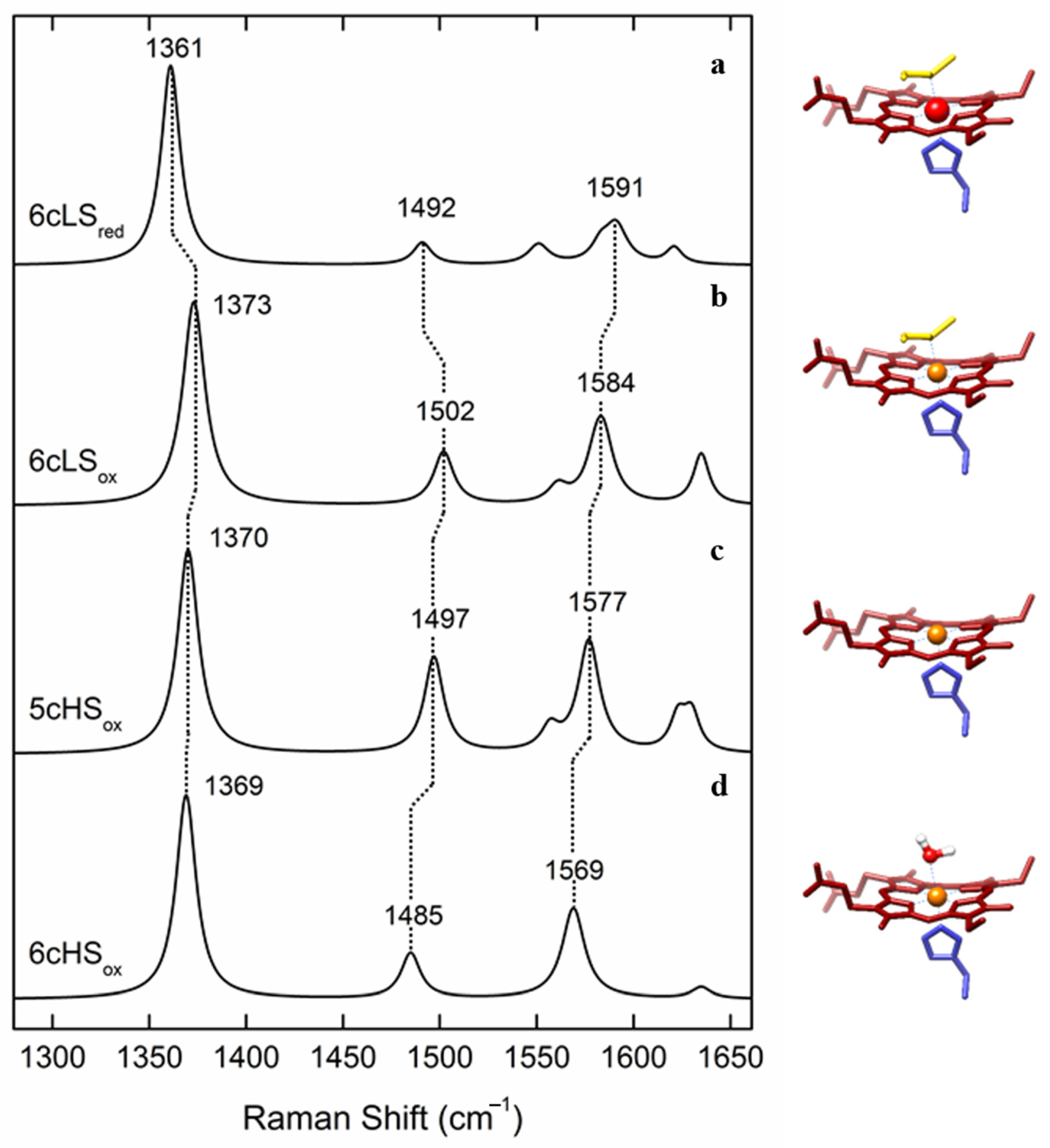
More recently, SERR spectroscopy provided a novel platform for investigations of HCOs under conditions that can mimic some basic features of their natural environment. Since these enzymes exert their function integrated into a phospholipid bilayer under restricted mobility, directionalized ET from electron donor to LS heme and binuclear site, and influence of strong interfacial electric fields, the immobilization onto biocompatible electrodes mimics far better these conditions than the solution studies [
16]. Furthermore, SERR potentiometric titrations represent a powerful alternative to common methods for the determination of the
E0 value, which is a prerequisite for understanding the ET pathway in these enzymes. The determination of
E0 of the individual heme groups of HCOs by conventional electronic absorption titrations is hampered by strong overlap** of the spectra of individual hemes and by complex cooperative effects that modulate the electroprotonic energy transduction [
16]. To that end, an
aa3-type quinol oxidase (
aa3 QO) that contains the catalytic and one cofactor-free non-catalytic subunit (
Figure 4a) was attached to detergent-coated Ag electrodes by spontaneous adsorption. A comparison of RR and SERR spectra of the enzyme in solution and adsorbed states revealed that the enzyme preserved its native structure upon immobilization (
Figure 5a). Deconvolution of the (SE)RR spectra by component analysis (
Figure 5b,c) allowed separation of the contributions from the LS and the HS hemes based on their respective ν
3 and ν
C=O modes, among more than 40 vibrational modes originating from the two hemes [
16]. The potential dependence of these spin- and redox-state-sensitive marker bands allowed for determination of midpoint redox potentials of hemes
a (
E0 = 320 mV vs. NHE) and
a3 (
E0 = 390 mV vs. NHE), which reveal a reversed order of reduction compared to mitochondrial-like HCOs. This suggests a distinct mechanism of electroprotonic energy transduction in
aa3 QO in comparison with, e.g., C
cO (
Figure 5d). A downhill ET is already guaranteed by the order of the midpoint redox potentials at the onset of enzyme reduction, indicating that this enzyme does not require a complex network of cooperativities to ensure exergonicity. Another SERR study employed a more complex, pentahemic
cbb3-type HCO, anchored to nanostructured Ni-NTA-coated Ag electrodes and further embedded into a lipid bilayer that mimics the natural membrane, which was catalytically active within the electrode construct [
64]. This strategy, which allows the control of the enzyme orientation that can be probed by SEIRA spectroscopy, was first developed in the studies of
aa3-type HCO [
17,
65,
66]. SERR potentiometric titrations of the whole
cbb3-type HCO complex and of the two individually expressed non-catalytic subunits, indicate that the dihemic subunit can be considered redundant for ET and catalysis, supporting the hypothesis that it plays a role in oxygen sensing. SERR also helped clarify the poorly understood coupling between heme reduction and proton translocation in HCOs and in particular the involvement of heme
a3 propionates in the proton pum**. The analysis was based on the protonation dependent CH
2 propionate bending modes that have been detected by H
2O-D
2O (SE)RR difference spectroscopy. This allowed individual assignment of all four heme propionates. The data support the hypothesis that heme
a3 propionates act as possible proton loading sites in HCOs [
67].
IR difference spectroscopy was employed to specifically probe the heme propionate protonation events using a C
cO mutant with
13C labeled propionates to distinguish its signal from possible concomitant changes of CO stretching modes of carboxylic acid groups of side chains [
18]. In such a way, differential IR spectra, employing isotopically labeled ligands or different redox states of the protein (e.g., oxidized minus reduced), provided further structural details on active site conformations in HCOs and specific protonatable sites [
68,
69]. It was demonstrated that NO binding to
cbb3, which also efficiently reduces NO to N
2O, exclusively occurs via HS heme
b3, and not via Cu
B as suggested [
70]. Step-scan IR spectroscopy, using Cu
B-CO adduct as a probe, showed that the protonation events in the binuclear site of
ba3-type HCO involve Tyr residues [
71]. Moreover, electrochemical redox titrations of
aa3- and
caa3-type HCOs, performed using IR spectroscopy in the transmission and the ATR mode, helped identify the redox-sensitive bands originating from heme groups, their ligands, amino acid residues and/or protein backbone. This strategy allowed for tentative assignment of the redox transitions of the individual redox centers [
36,
68,
72]. A similar approach, in which electrochemistry coupled to redox induced IR difference spectroscopy was used to address the
E0 of the heme
b3 in
cbb3 HCOs, revealed relatively low midpoint redox potential of this HS heme. It was rationalized in terms of a unique coordination scheme involving hydrogen bonding between the His ligand of the heme
b3 and a highly conserved glutamic acid, which participate in coupled electron and proton transfer steps and facilitate ET between LS and HS hemes during turnover [
73].
Nitric oxide reductase (NOR) catalyzes the two-electron reduction of NO to N
2O at the di-nuclear heme
b3-Fe
B center. Similarly to the active site of HCOs, the heme
b3 is in the HS state, while Fe
B is a non-hemic iron atom. NORs in addition house
c-type and
b-type LS hemes. RR spectroscopy provided evidence about NO reduction by monitoring the events that occur upon NO binding to the catalytic
b3-type heme. Specifically, formation of the N-N bond and the fate of the proximal His-heme
b3 bond along this process, as well as the recovery of the
b3-O-Fe
B state, have been disentangled by RR spectroscopy [
74].


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}