3.1. Experimental Evidence on the Coordination Ability of [(ppy)Au(IPr)]2+
The catalytic activity of [(ppy)Au(IPr)Cl]Cl (
1) in the 3-hexyne hydration reaction has shown a not completely satisfactory efficiency of this complex [
40].
However, in an attempt to clarify key aspects of the mechanism and to characterize and isolate the reaction intermediates of the pre-equilibrium step of the alkyne hydration reaction mechanism, namely the complexes [(ppy)Au(IPr)(3-hexyne)]
2+ (
2), [(ppy)Au(IPr)H
2O]
2+ (
3), [(ppy)Au(IPr)OTf]
+ (
4), and [(ppy)Au(IPr)BF
4]
+ (
5), the reactivity of
1 towards the addition of AgBF
4 or AgOTf in different solvents, in the presence of 3-hexyne or water was studied. This exploration allowed to gain useful insight into the coordination ability of the [(ppy)Au(IPr)]
2+ fragment towards the different species present in the catalytic conditions. Relevant findings are briefly described below (see
Scheme 1). The starting complex (or pre-catalyst)
1 coordinates the chloride ions in the first and second coordination shells. Cl
− must be removed in order to coordinate and activate the 3-hexyne substrate. Experimentally, this step requires the addition of silver salts, and therefore AgOTf and AgBF
4 were used to remove the Cl
− ions in different conditions (
Scheme 1). [(ppy)Au(IPr)H
2O](BF
4)
2 (
3BF4) and [(ppy)Au(IPr)BF
4]BF
4 (
5BF4) complexes were generated in a NMR tube by reacting
1 with 2 equiv of AgBF
4 in non-anhydrous CD
2Cl
2 (for experimental study details see
Supplementary Materials, Scheme S1, Figures S1–S11). From
1H,
13C,
1H-COSY,
1H-NOESY,
1H,
13C-HMQC NMR, and
1H,
13C-HMBC NMR spectroscopies all proton and carbon resonances belonging to the different fragments of both complexes were assigned. The two complexes are present in about equal amount (52% of
5BF4) and monitoring the chemical exchange between the two resonances of proton H1 by qualitative
1H-EXSY NMR experiments allowed to extract the rate constant (k
obs) for their interconversion, k
obs was found to be 1.0 s
−1.
In order to synthesize the complex [(ppy)Au(IPr)(3-hexyne)](BF4)2 (2BF4), 10 equiv. of 3-hexyne were added to the CD2Cl2 solution containing 3BF4 and 5BF4, but no variations of the resonances belonging to both complexes were observed, thus indicating that no formation of alkyne complex 2BF4 was attained. On the contrary, the addition of 5 equiv. of water quantitatively shifted the 3BF4/5BF4 equilibrium towards the former.
Then, we decided to try to synthesize [(ppy)Au(IPr)H
2O](OTf)
2 (
3OTf) and [(ppy)Au(IPr)OTf]OTf (
4OTf) complexes by following the same procedure, but the formation of [(ppy)Au(IPr)Cl]OTf (
6OTf) was obtained (
Scheme 1). The addition of 4 equiv. of AgOTf to a solution of 1 in acetone at 50 °C overnight gave the formation of a mixture of
6OTf,
4OTf, and
3OTf in 0.22, 0.70, 0.08 ratio (see
Supplementary Materials for details). Analogously in this case, the addition of 10 equiv. of 3-hexyne to a CD
2Cl
2 solution of the mixture did not change its composition, while the addition of water gave the formation of a mixture of
6OTf and
3OTf.
For a direct comparison with the mixture of
3BF4/
5BF4 in dichloromethane, the NMR spectrum of the mixture
4OTf/
3OTf was recorded in the same solvent. A similar composition of both mixtures was observed indicating a similar coordination behavior of BF
4− and OTf
− towards the gold fragment (see
Supplementary Materials).
In summary, the species
2 was not detected in our NMR experiments when BF
4−, OTf
− or water are present into the mixture. In addition, BF
4− and OTf
− showed the same coordinating ability towards [(ppy)Au(IPr)]
2+ and slightly less than water. To date, only the water-containing species
3BF4 can be isolated and characterized, consistently with the highly reactive character of the catalyst-substrate complex during the catalysis [
39].
These results contrast with what found for instance for the [(NHC)Au]
+ (NHC = 1,3-dimethylimidazol-2-ylidene) fragment, where 3-hexyne and OTf
− are more coordinating than water and BF
4− [
43,
44,
45,
46,
47,
48]. As a partial confirmation, Glorius and coworkers have isolated a similar compound, [(ppy)Au(PPh
3)BF
4]
+, in which the BF
4− ion is coordinated in the inner sphere instead of water [
62].
These experimental counterintuitive results and challenges have been addressed computationally in this work in an attempt to rationalize them and, eventually, to suggest how to improve the catalytic efficiency of this type of Au(III) complexes.
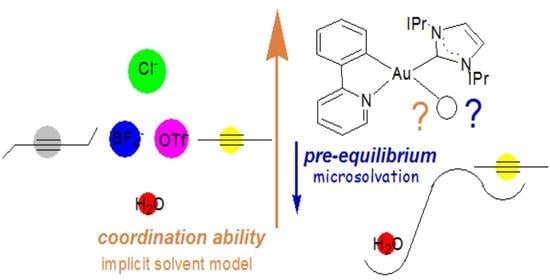
3.2. Coordination Ability of the [(ppy)Au(IPr)]2+ Fragment
To study the coordination ability of the [(ppy)Au(IPr)]
2+ fragment, the bonding energies of X (X = Cl
−, BF
4−, OTf
−, H
2O, 2-butyne and 3-hexyne) have been evaluated. We have performed calculations of electronic energy (∆E), enthalpy (∆H) and Gibbs free energy (ΔG) change for the following reaction:
As the X ligands are concerned, we selected all the experimentally employed species. In addition to 3-hexyne, also 2-butyne was examined, which is usually considered as a model alkyne in computational investigations. The results are summarized in
Table 1.
The bonding free energies (∆G) span a range of −0.4/−44.0 kcal/mol and, as a general trend, we can immediately see that the coordination ability decreases in the order of Cl
− > OTf
− > 3-hexyne > 2-butyne ≈ BF
4− > H
2O. Note that the same qualitative trend can be found considering the ∆E and ∆H values. Remarkably, ∆G values for OTf
−, BF
4−, 3-hexyne and 2-butyne are very close, in the range of −12.5/−17.5 kcal/mol, thus suggesting that they should have a similar coordination ability towards [(ppy)Au(IPr)]
2+or, rather, that the [(ppy)Au(IPr)]
2+ fragment should be poorly selective towards these four ligands, irrespective of their different charge. The Cl
− strongest coordination ability is in full agreement with the difficulty experimentally encountered when attempts are made to remove the chloride from the first coordination shell of the Au(III) complex. By contrast, removal of Cl
− from L-Au-Cl by AgX is experimentally easier [
47]. Results reported in
Table S1 in the
Supplementary Materials show however that free energy for chloride binding to Au(III) is very similar to that to Au(I) (−44.0 vs. −43.9 kcal/mol, respectively). Then, other effects should be taken into account to rationalize this finding. On the computational side, for instance, the continuum implicit solvent model we are using here could not be completely satisfactory for comparing Au(III)-Au(I) coordination ability to chloride at the actual experimental conditions (for a discussion on this issue, see
Section 3.4).
On comparing the coordination ability of BF
4− and OTf
− to the metal center, a difference of only 5.0 kcal/mol is calculated, with OTf
− being more coordinating. This relatively low value is unexpected on the basis of the commonly recognized, both theoretically and experimentally, higher coordination ability of OTf
− compared to BF
4−. However, this finding is fully consistent with the experimentally observed similar coordination ability of BF
4− and OTf
− towards the [(ppy)Au(IPr)]
2+ fragment, which is only slightly shifted in favour of the triflate ion. We can surmise that the large steric hindrance due to the two isopropylphenyl groups of IPr is responsible for a larger destabilization of the Au(III)-OTf bond than the Au(III)-BF
4 one caused by the larger size and less “spherical” symmetry of OTf
− compared to BF
4−. As a consequence, the coordination ability of OTf
− decreases in the [(ppy)Au(IPr)X]
+ complex, although remaining slightly more coordinating than BF
4−. However, the decreasing contribution of the steric hindrance to the OTf
− bonding energy could be counteracted by an additional effect still due to the IPr and ppy ligands, namely the long-range noncovalent interactions, which are demonstrated to play an important role in these large dimension systems where high polarization of the ligands fragments is expected [
63,
64]. In the optimized structures of the [(ppy)Au(IPr)X]
+/2+ (X = Cl
−, OTf
−, BF
4−, 3-hexyne, 2-butyne, and H
2O) complexes which are compared in
Figure S12 in the
Supplementary Materials, the nearest contacts between the ligand X and the hydrogens from the ppy ring and the isopropylphenyl groups of IPr are highlighted.
Precisely in order to investigate this issue, the coordination ability trend in a [(ppy)Au(NHC)]
2+ (NHC = 1,3-dimethylimidazol-2-ylidene) fragment, with a simplified IPr ligand (the two isopropylphenyl moieties are replaced by methyl groups), commonly used in computational studies on IPr-based gold complexes, is analyzed. A comparative analysis of the experimental vs. model fragment coordination ability is presented in the
Supplementary Materials (
Table S1,
Figures S13 and S14). The bonding energies of the [(ppy)Au(NHC)]
2+ fragment generally increase for all the X ligands with respect to those of [(ppy)Au(IPr)]
2+ (by 6-15 kcal/mol) and the [(ppy)Au(NHC)]
2+ fragment is found to be more selective towards OTf
− than BF
4− (the difference in ∆G increases from 5 kcal/mol in [(ppy)Au(IPr)]
2+ to 10 kcal/mol in [(ppy)Au(NHC)]
2+). Overall, these results suggest that the most important contribution to the OTf
− bonding energy in the [(ppy)Au(IPr)]
2+ fragment is much more likely the destabilizing steric effect of IPr.
Focusing on neutral ligand series, as a balance between the steric effects due to the IPr ligand and the stabilizing effects of long-range noncovalent interactions between alkyne and IPr and ppy, both the alkynes are found to be more strongly coordinating to Au than water, with larger size 3-hexyne coordinating better than 2-butyne (see
Table 1), thus indicating that the steric hindrance should not be the only effect that contributes to the selectivity of the metal towards the considered neutral ligands. Remarkably, H
2O exhibits the smallest calculated bonding free energy in both the [(ppy)Au(IPr)H
2O]
2+ and [(ppy)Au(NHC)H
2O]
2+ complexes (
Table 1,
Table S1 and
Supplementary Materials). This finding, which is in strong disagreement with the experiment, suggests that the [(ppy)Au(IPr)]
2+ complex under study would be more π-philic than oxophilic, at variance with common assumption in the literature that gold(III) catalysts do have a strong oxophilic nature, whereas gold(I) catalysts do not [
17,
65,
66,
67]. A comparison with the coordination ability of a [(NHC)Au]
+ fragment towards X = Cl
−, OTf
−, BF
4−, 3-hexyne, 2-butyne, H
2O has been also reported in the
Supplementary Materials (
Table S1,
Figure S15), showing that, analogously, H
2O is the weakest ligand in the whole series. In conclusion, due to the steric hindrance/dispersion interaction balance between X and IPr, the [(ppy)Au(IPr)]
2+ fragment is found to be less selective towards the different ligands, with OTf
− and BF
4− showing very similar coordination strength, thus rationalizing the experimentally observed equilibrium between
4OTf and
3OTf and, analogously, between
5BF4 and
3BF4. On the contrary, this factor cannot explain the stability of the water-containing species
3BF4 which could be experimentally isolated and characterized and vice versa the failure in isolating the alkyne complexes. The coordination ability has been evaluated using an implicit continuum solvation model which may be not fully satisfactory to establish an accurate scale of coordination ability at the actual catalytic conditions. Before addressing this issue, in the following section we investigate the effect of the ancillary ligand on the coordination ability of the metal fragment towards alkynes and water.
3.3. Ligand Effect on the Coordination Ability: Alkynes vs. Water
To shed light into the above intriguing results for neutral ligands, where H2O results a weaker coordinating ligand than alkynes, the ancillary ligand influence on the Au(III) coordination ability is now analyzed.
Among suitable ancillary ligands, we selected the
bis-cyclometalated [(C^N^C)Au]
+ (C^N^C = 2,6-
bis(4-
tBuC
6H
3)
2 pyridine dianion) and the monocyclometalated [(ppy)AuCl]
+ Au(III) complexes. In particular, alkyne complexes with [(C^N^C)Au(III)]
+ have been experimentally observed and characterized by Bochmann et al. with different internal alkynes [
68,
69]. Theoretical studies on the [(C^N^C)Au(III)]
+ alkyne and carbonyl complexes have also been performed previously by some of us [
39,
70]. Instead, the [(ppy)AuCl]
+ complex, where a Cl
− replaces the IPr ligand in our reference complex, allows for a direct comparison with [(ppy)Au(IPr)]
2+, where the X interactions with IPr are switched off. In addition, the [(ppy)Au(NHC)]
2+ complex has been considered to test the commonly used procedure of modelling the IPr with NHC in computational studies and the Au(I) complex, [(NHC)Au]
+ is examined, for a comparison with Au(III). The optimized structures of [(C^N^C)AuX]
+ and [(ppy)AuClX]
+ (X = H
2O, 2-butyne, 3-hexyne) are shown in the
Supplementary Materials (
Figure S16).
Analogously to [(ppy)Au(IPr)]
2+, the bonding free energies of X (X = H
2O, 2-butyne, 3-hexyne) have been calculated for the following reactions:
From
Table S2 and
Figure 1 we can observe that, although the coordination ability of Au(III) is quantitatively strongly affected by the ligands, the coordination ability trend is the same for all the fragments, namely 3-hexyne > 2-butyne > H
2O. In particular, for a given X (X = H
2O, 3-hexyne, 2-butyne), the coordination ability follows the trend [(C^N^C)Au]
+ > [(NHC)Au]
+ > [(ppy)Au(NHC)]
2+ > [(ppy)AuCl]
+ > [(ppy)Au(IPr)]
2+. One dominant factor influencing the strength of the Au-X interaction might be the trans influence of the ancillary ligand [
68]. In [(C^N^C)AuX]
+ the X ligand feels only the relatively weak effect of a trans pyridine, whereas in [(ppy)AuClX]
+, [(ppy)Au(NHC)X]
2+ and [(ppy)Au(IPr)X]
2+ the X is trans to a phenyl group with a much stronger effect. Notably, only Au(III) in [(C^N^C)Au]
+ can coordinate alkynes better than the [(NHC)Au]
+ fragment. In addition, these results suggest that Au(III) coordination ability does not depend on the charge of the complex: for example, [(C^N^C)Au]
+ coordinates to X more strongly than [(ppy)Au(NHC)]
2+ with 2+ charge which, in turn, coordinates to X more strongly than [(ppy)AuCl]
+. Again, these findings seem to do not support a commonly encountered postulate in the literature according to which Au(III) species are more oxophilic in nature whereas Au(I) species show a more π-philic property [
17,
65,
66], since, within the considered auxiliary ligands set, both Au(III) and Au(I) show a stronger coordination ability towards alkynes than water, at least within an implicit solvent model. Finally, on comparing [(ppy)Au(NHC)]
2+, [(ppy)AuCl]
+ and [(ppy)Au(IPr)]
2+, replacing of IPr with NHC or Cl
− has a beneficial effect on coordination ability, thus reasserting a sizable role of IPr steric interactions with X.
3.4. Substitution of H2O With the Substrate: The Pre-Equilibrium Step
From the above study, water clearly emerges as the weakest ligand in the whole series and significantly weaker than alkynes. All the studied Au(III) complexes are important species in catalytic alkyne nucleophilic addition reaction. The first step of the reaction mechanism is represented by the pre-equilibrium, where the substrate (alkyne) replaces the anion in the initial complex, binds to the gold center, and is activated for the nucleophilic attack. However, in the alkyne hydration reaction, where H
2O is the nucleophile, the anion substitution by the water molecule, i.e., formation of the water adduct from the corresponding initial complex, has been shown to be both thermodynamically and kinetically favored with respect to the formation of alkyne complex, in agreement with the experimental evidence (see also Introduction and
Scheme 1 for details) [
40]. Therefore, starting from the experimentally isolated and characterized
3, we will explore the pre-equilibrium step consisting of H
2O substitution by the alkyne and rationalize the reason why
2 is not experimentally observed. The experimental conditions of the alkyne hydration reaction need to be clearly modelled beyond the implicit solvation model we have employed above. In a reaction environment where anionic ligands are not close to the Au(III) catalytic complex (for instance, using highly polar solvents or very weakly coordinating anions) the present species can be additional water, alkyne and/or high polarity solvent molecules. To model such reaction conditions, calculations of the initial complex (IC) and reactant complex (RC) using the Au(III) model complex and 2-butyne have been performed in an attempt to study the pre-equilibrium step of the hydration reaction mechanism. The IC is represented by the Au(III)-H
2O model complex with 2-butyne in the second coordination sphere, whereas the RC is the Au(III)-2-butyne model complex with H
2O in the second coordination sphere. To account for the experimental conditions, the IC and RC calculations have been performed including: i) an additional water molecule to model traces of water or water solvent effect; ii) a GVL molecule to model the highly polar aprotic solvent effect actually used in the experimental study. We should remark that these calculations have been performed with exactly the same computational protocol as that used for the coordination ability study, namely by including the continuum solvent model (COSMO) with dichloromethane as the solvent, for a direct comparison with data in
Table S1 in the
Supplementary Materials. The results are summarized in
Figure 2, where the energy profiles for the pre-equilibrium step have been shown.
Interestingly, in both cases IC is found to be more stable than RC, thus indicating that water is a stronger ligand than 2-butyne if we include in our model the microsolvation effect, which is more closely related with the actual experimental conditions. It is eye-catching that the IC stabilization is due to an explicit interaction of the coordinated H
2O with a solvent molecule, namely that only microsolvation can account for “oxophilicity” of gold(III) which could not be found using an implicit solvent model (COSMO calculations). The same result has been also found in the gas phase (no solvation model), where the aquo complex IC is more stable than the 2-butyne complex RC by 6.4–4.5 kcal/mol (see corresponding
Figure S21 in the
Supplementary Materials). Transition state calculations for the IC → RC process show that the free energy activation barrier is 6.0 kcal/mol for H
2O and 4.2 kcal/mol for GVL. However, the reverse process RC → IC requires a lower barrier (only 2.0 kcal/mol for GVL and 3.7 kcal/mol for H
2O). Our calculations clearly point out that IC is thermodynamically more stable than RC, which rationalizes the difficulty to isolate and characterize the alkyne complex in the experimental conditions: anytime the complex binds to the alkyne, it is substituted by the H
2O.
The presence of a polar molecule (H2O or GVL) which is able to establish a hydrogen interaction with water in ICH2O or ICGVL polarizes the water O-H bond, increasing the partial negative charge on the oxygen atom which becomes more coordinating toward the metal center. The lower activation energy barrier obtained when the solvent is GVL, compared to water, suggests that the “activation” of the O-H bond is prevalently caused by the hydrogen bond strength. Indeed, in ICGVL structure, the water O-H distance involved in GVL hydrogen bond is 1.029 Å, whereas in ICH2O structure the corresponding O-H distance is 1.026 Å, thus indicating that GVL is more efficient in “activating” H2O to an incipient “OH−”.
A direct comparison with the Au(I) catalyst, [(NHC)Au]
+ is reported in
Figure 2. The most noticeable difference relies on the more stable RC compared to IC. Therefore, in the Au(I) case, water is less strong than 2-butyne as ligand, and an equilibrium shift towards the RC species is suggested. The partial negative charge on the oxygen atom of the water O-H bond induced by microsolvation appears to be less stabilized by gold +1 charge than +3, as one could expect. A more realistic description of the experimental conditions would however require a sample of the conformational space with inclusion of a large number of explicit water or GVL molecules. These calculations are outside the scope of the present paper. On the other hand, our simple model allows for a detailed microscopic rationalization for experimental findings which interestingly suggests that microsolvation could be very important for the pre-equilibrium step of the [(ppy)Au(IPr)]
2+–catalysed hydration of alkynes and which calls for further investigations on this issue in other Au(III)-catalyzed reaction types.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}