2.1. Possible Low Energy Structures of the Four Diastereomers
While chemical intuition and prior experimental structural evidence have been used in some reports, such as in the IR spectroscopic study of (Ser
8Cl
2)
2− [
33] and (HSer
8)
+ [
23], increasingly a global search for suitable structures has been carried out by the laser-mass spectrometry community using some MD simulation program packages, such as Macromodel by Schrödinger [
34] and DFTB+, which utilizes the DFT-based tight binding method [
35,
36]. For example, Poline et al. applied this approach to investigate the IR signature of several homo- and heterochiral protonated amino acid dimers [
37]. We chose to use the conformer-rotamer ensemble sampling tool (CREST), developed by Grimme and co-workers [
38]. CREST has been applied extensively and successfully to rotational spectroscopic studies of the conformational landscapes of mid-sized neutral organic molecules and their clusters [
39,
40]. Furthermore, it was also benchmarked for its ability to correctly predict protonation sites [
41].
In some recent rotational spectroscopic studies of fluoroalcohol trimers and tetramers [
42,
43], it was recognized that monomeric conformations which are not stable in their isolated form, may become the main or even the only subunits in larger aggregates, highlighting the importance of extensive sampling of the conformational space. While each CREST run already has 12 built-in MD runs, to cover as much conformational space as possible, multiple CREST runs with the same or different starting geometries were added in the current study. One reason is that while performing conformational analyses, we noticed that a high torsional barrier exists in all species, which locks the OH group of the carboxylic group in either a cis or trans configuration relative to the C=O group. A similar phenomenon was reported in the rotational spectroscopic studies of tetrahydro-2-furoic acid [
44] and its dimer [
11]. In a single CREST run, redundant structures are removed by the program itself. Since many conformers found in different CREST runs could also be the same, we used a Python program to calculate the root-mean-square deviation (RMSD) of atomic Cartesian coordinates for the obtained conformations. A RMSD value of zero indicates identical conformations, whereas the larger the value, the more unalike the two conformations are. By trial and error, a RMSD threshold of 0.6 was used.
While some have chosen to directly use the xTB energy to choose structural candidates for further DFT optimization, our experience with neutral aggregates suggested that the xTB energy ranking may be misleading in some cases [
40]. An initial survey of the single-point energies of the CREST geometries was performed and the result suggested that such energy values were still too far from the final ones for them to be used as credible discriminators. Because of the large number of initial CREST candidates, we decided to add a few additional fast computational steps to obtain a more reliable energy ranking so that we could properly select the low energy structures for final geometry optimization and harmonic frequency calculations. These include (1) CREST output; (2) DFT optimizations with a relaxed convergence criteria of initial CREST candidates at the revPBE-D3/def2-SVP [
45] level, with the empirical D3 dispersion correction [
46,
47]; and (3) a single-point energy evaluation at the B3LYP-D3/def2-TZVP level of the optimized structures in step (2).
Figure 1 shows the relative energies for 42
S,
S-HSerAsn
+ structures at the three different calculation stages and their correlation with the B3LYP-D3/def2-TZVP energies after geometry optimization using Molpro [
48,
49]. It is apparent that GFN2-xTB generally underestimates the relative energies of the CREST conformers, resulting in “overcrowding” of the initial conformer ensemble within a given energy window, making it difficult to carry out the selection. The revPBE relative energies, on the other hand, tend to be overestimated. Finally, single-point B3LYP calculations of the revPBE-optimized structures yield very reliable estimates of the B3LYP-D3/def2-TZVP energies after geometry optimization, save for a few outliers. This combined B3LYP-D3/revPBE-D3 step is therefore crucial in assessing the conformer ensembles, since it strikes a very attractive balance between computational effort and accuracy without requiring the full B3LYP-D3 optimizations.
For the four chiral diastereomers of interest, the final optimization and frequency calculations of the structures selected within an energy window of ~10 kJ mol
−1 from the global minimum of each species were performed at the B3LYP-D3BJ/def2-TZVP level using Gaussian 16 [
50]. The energetic properties and the associated Boltzmann factors are summarized in
Tables S1–S4 (
Supplementary Material) for
S,
S-HSerAsn
+,
R,
S-HSerAsn
+,
S,
S-HValAsn
+, and
R,
S-HValAsn
+, respectively. The single-point energy calculations of all the above structures with the inclusion of a solvent polarizable continuum model (PCM) implemented in Gaussian 16 [
50] are also included in the corresponding Tables. Unsurprisingly, the zwitterionic (ZW) form of these binary clusters is strongly preferred with the inclusion of a solvent.
The global minimum structures of each species are presented in
Figure 2, as well as the second most stable structures of
S,
S-HSerAsn
+ and
R,
S-HSerAsn
+. In addition, to visualize the full range of sidechain orientations that each dimer species realizes within the 10 kJ mol
−1 window, important atoms are color coded. The matching dots are then used to indicate the positions of these specific atoms in all structures beyond the explicitly depicted minima after alignment by RMSD minimization of all non-hydrogen atoms. All these structures have the protonation site on Ser or Val, while Asn takes on the neutral or ZW form.
2.2. Different Noncovalent Binding Topologies
In the previous rotational spectroscopic studies of the conformational landscapes of monomeric, neutral amino acids [
51], it was recognized that α-amino acids with a nonpolar sidechain such as Val, typically present in only two dominant conformers stabilized by either a bifurcated N–H⋯O=C hydrogen bond with a cis-COOH configuration or a N⋯H–O hydrogen bond [
31]. With the presence of polar sidechains, the number of conformers with similar energies tends to increase dramatically. For example, seven conformers were observed for neutral Ser [
30]. Interestingly only one main conformer of Asn was identified in the previous rotational spectroscopic study [
32], an exception to the rule. Asn utilizes both cis- and trans-COOH configurations in the binary species studied here, although it can take on neutral, protonated, or ZW forms, a point which will be further discussed in
Section 2.4. Generally, the greater conformational diversities of the Ser monomer versus Val also seem to be reflected in their respective protonated binary species (
Figure 2), where the sidechain of the Ser subunit has the tendency to occupy far more different regions in the above dimers than the sidechain of Val.
To better appreciate structural diversities in the homo- and heterochiral HSerAsn
+ and HValAsn
+ dimers, we divided these isomers into five types based mainly on their key intermolecular interaction difference, the subunit conformations and whether the subunits take on the ZW form or not. These include Type I, II, and III of the protonated form where the IR band signatures look very similar within each type but different from each other, then the ZW form, and finally some minor structures labelled as ‘Other’ which do not belong to the previous four categories. These labels are also listed in
Tables S1–S4 for all four binary species. Based on the predicted relative free energies, Type I, II, and III of HValAsn
+ make up almost all the population, whereas those labelled as ‘Other’ contribute very little. In contrast, for HSerAsn
+, those labelled as “Other” have a higher contribution. This outcome is expected since the polar sidechain of Ser offers more potential binding sites with Asn.
In
Figure 3, the geometries of the most stable isomers of each type of
S,
S-HSerAsn
+ and
R,
S-HSerAsn
+ are provided, where the dominant intermolecular hydrogen bonds are also indicated, together with their relative energies. Since the relative stability ranking of the isomers might change based on ΔE or ΔG, we used ΔG for ranking in the remainder of the paper. Type I structures contain two intermolecular hydrogen bonds connecting the NH
3+ group of Ser with the nitrogen atom and the carbonyl oxygen atom of Asn. Type II isomers have an intermolecular hydrogen bond between the Ser NH
3+ group and the carboxyl O atom of Asn and another from the Ser OH group to the carbonyl O atom of Asn. At the same time, the carboxyl OH group of Asn maintains an intramolecular hydrogen bond with its own nitrogen atom where the backbone dihedral angle of the Asn subunit needs to be turned to facilitate this intramolecular hydrogen bond, leading to a slight destabilization of the whole structure. Type III isomers are unique compared to the other three because the protonation site is on the Asn subunit instead of Ser. In Type III isomers, the nitrogen atom of Ser forms an intermolecular hydrogen bond with the NH
3+ group of Asn, while an intramolecular hydrogen bond is formed between the carbonyl O atom of Ser and its NH
2 group. As the Asn subunit takes on the ZW form, two binding motifs are identified for HSerAsn
+: ZW1 where the polar sidechain of Ser is not involved in the intermolecular interaction and ZW2 where the Ser sidechain is involved. By changing how a subunit approached the H atom(s) of the NH
3+ group of Ser to form the respective hydrogen bonding interactions, three isomers having Type I structures were found, and two for each of Type II, III and ZW2.
Similarly, the four types of binding topologies of
S,
S-HValAsn
+ and
R,
S-HValAsn
+ are depicted in
Figure 4. Type I and II structures of the HValAsn
+ species contain very similar intermolecular hydrogen bonds as in the corresponding Type I and II of the HSerAsn
+ species, respectively. The exception is that in the Type II of the HValAsn
+ species, the COOH of Val acts as a proton donor instead of the OH group of Ser in the HSerAsn
+ species case. This is not surprising since Val does not have an alcohol OH group. Type III of the HValAsn
+ species utilizes the NH
3+ of Val as the proton donor to the carbonyl and carboxylic O atoms of Asn, a very different binding topology compared to that of the HSerAsn
+ species. Unlike HSerAsn
+, only one binding motif is observed for the ZW structures since their nonpolar sidechain is not a competitive intermolecular hydrogen bond donor candidate compared to the OH group of Ser.
Figure 5 visualizes the relative free energies of all the final structures within the 10 kJ mol
−1 window for the four dimer species. The data points in each trace are coloured according to their binding topologies: Type I, II, III, ZW and ‘Other’. In the case of the HserAsn
+ species, the lowest energy structures of the homo- and heterochiral complexes appear to be the same type, Type ZW. For the HvalAsn
+ species, the lowest energy structures of the homo- and heterochiral complexes also appear to be the same type, Type I.
2.3. Comparison of the Experimental and Theoretical IRMPD Spectra
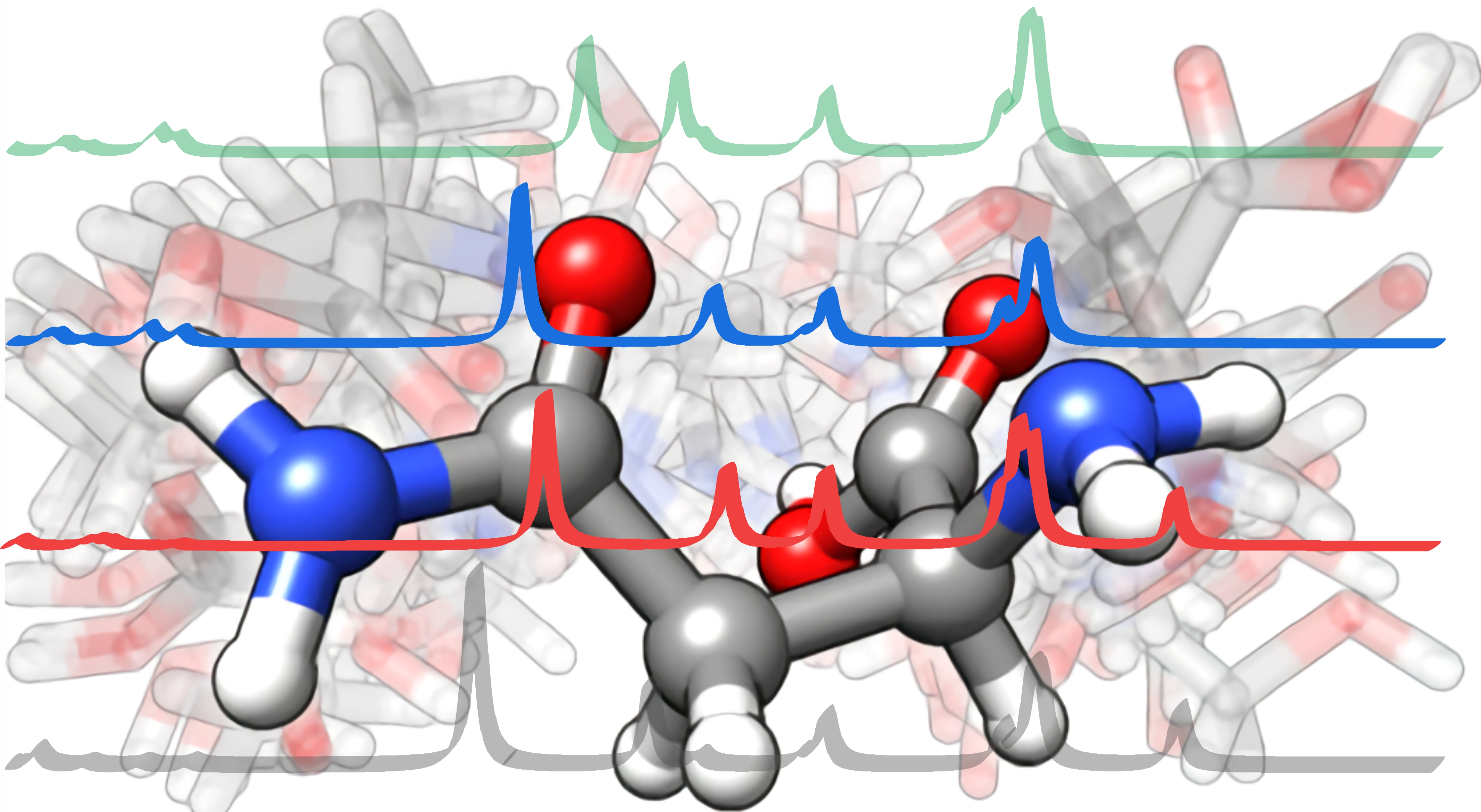
In
Figure 6, the experimental IRMPD spectra of the homo- and heterochiral HSerAsn
+ dimers are compared with the theoretical, individual IR spectrums of the most stable isomers of the five relevant types, while detailed comparisons between the experimental and theoretical spectra of each type are summarized in
Figures S1 and S2,
Supplementary Material. Both homo- and heterochiral experimental spectra show four clear band features, labelled as B, C, D, and E, in the above 3200 cm
−1 region. Experimentally, there is also a small shoulder band to the lower wavenumber side of D which only becomes obvious at a higher radiation energy of 15 mJ. Based on the comparison between the experimental and simulated spectral features, one can rule out contributions of Type II, ZW1 and ZW2 for both
S,
S-HSerAsn
+ and
R,
S-HSerAsn
+. Below, we focus on the remaining Type I and III structures.
For S,S-HSerAsn+, Type I, #2 provides the best overall agreement with the experimental IR pattern including the small shoulder band next to D and in terms of the predicted energetic preference, i.e., the most stable one among the remaining Type I and III. The other four isomers, i.e., Type I, #8 and #12, and Type III, #5 and #9, all show features consistent with C, D, and E. It appears that the contribution from them can be used to explain the broadening observed in the experimental D band, and also the much broader B band where the corresponding calculated B bands of these isomers extend over some tens of wavenumbers.
In terms of the vibrational assignment, the highest frequency E band corresponds to the free OH stretch of Ser which is not involved in the noncovalent intermolecular interaction; the D band can be related to the carboxyl OH stretches of the Ser and Asn subunits. Neither of the carboxyl OH stretches are involved in the intermolecular hydrogen bonds and are predicted to be close in their frequencies. The “shoulder” peak (next to D) at ~3550 cm
−1 corresponds to the asymmetric stretching motions of the sidechain NH
2 group of Asn. The C band contains the symmetric stretching information of the sidechain NH
2 group of Asn, as well as the asymmetric stretching motion of the proton acceptor NH
2 group of Asn, the intensity of which is lower and appears as a smaller “shoulder” in the predicted spectra. The B band is assigned to a collection of symmetric stretch motions of the proton acceptor NH
2 group of Asn and the asymmetric NH
2 vibrations in the proton bound NH
3+ groups of Ser, analogous to the previously published assignments [
24]. Note that the predicted symmetric NH
2 vibrations in the NH
3+ group of Ser fell into the “blind region” of our laser and could not be detected in the experiment.
In the region below 3200 cm
−1, the predicted IR bands are dominated by the stretching modes of NH
3+ which serves as a hydrogen bond donor in the dimers. These stretching bands exhibit typical characters such as large red shifts and a big enhancement in IR strength. Experimentally, it is well known in the jet-cooled high-resolution IR community that IR photons pumped into intermolecular hydrogen bonds tend to lead to severe predissociation broadening in the experimental IR spectra [
52]. In the current case, this results in a broad and featureless contour which is marked as band A, similar that which was observed previously [
24]. The backbone CH stretches from the Asn and Ser subunits are predicted to be in the 3000–3110 cm
−1 region and tend to be featureless because of the high density of the CH vibrational states [
22].
For R,S-HSerAsn+, the Type I, #5 and #7 isomers appear to provide the best agreement with the experimental observations, while the contribution from Type I, #10 and Type II, #6 and #12 is also present. The IR band assignments for A–E remain analogous to those of S,S-HSerAsn+. There are some minor experimental pattern differences from S,S-HSerAsn+: the D shoulder is less well resolved and some partially resolved structures in B are more obvious. These experimental observations can be explained by the smaller separation predicted for the D band and its shoulder band in Type I and III in R,S-HSerAsn+ and because the free energy gaps among the Type I and III isomers are smaller than the gap between Type I, #2 and the rest in the case of S,S-HSerAsn+, leading to a wider contribution of different isomers.
In
Figure 7, the experimental IRMPD spectra of homo- and heterochiral HValAsn
+ dimers are compared with the simulated individual IR spectrums of the most stable isomers of Type I, II, III and ZW1, while detailed comparisons between the experimental and theoretical spectra of each type are summarized in
Figures S3 and S4,
Supplementary Material. Type ZW2 is outside the 10 kJ mol
−1 free energy window.
The most obvious difference from the HSerAsn+ case, is the missing E band which belongs to the free OH of Ser, since HValAsn+ does not have such a free OH group. Based on the simulated IR spectral patterns in the region surrounding C, we can rule out the contribution of Type II and III. Both of them have a lower wavenumber band predicted next to C, which is not present in the experiment. The comparison of the simulated and experimental band gap between C and B’/B also allows one to discard the contribution of Type ZW1.
For S,S-HValAsn+, Type I, #1, #3 and #5 structures exhibit well-aligned C and D bands, i.e., very similar C and D frequencies among the three isomers, and more spread out B′/B bands, consistent with the experimental spectral appearance. The B and B’ of the Type I structures are assigned to the symmetric and asymmetric stretches of the hydrogen bonded NH3+ group of Val. The IR assignments of the C and D features are analogous to the HSerAsn+ case described above.
Similarly, for R,S-HValAsn+, Type I, #1, #2, and #5 structures contribute to the experimental IR spectra, as indicated by the good agreement with the experimental features. Contributions of Type II, Type III and Type ZW1 structures can be discarded for the same reasons provided above for S,S-HValAsn+.
2.4. Chirality Recognition and Kinetic Effects in the IRMPD Spectra
The above IRMPD spectral analyses show that while one could clearly discriminate among different types of structures for the four binary species studied here, only minor differences between the homo- and heterochiral species of HSerAsn+ and HValAsn+ were observed experimentally. Does this mean there is no or little chirality recognition in these homo- versus heterochiral species? In the following, we first address why the species which contribute to the experimental IRMPD patterns may not necessarily be the most stable structures predicted. Then we discuss some noticeable chirality recognition signatures and energetic differences in these systems, even if these are not reflected in the IRMPD features detected, and how one may tease out these signatures with modified experimental approaches.
Although the global minima predicted for both
S,
S-HSerAsn
+ and
R,
S-HSerAsn
+ belong to the ZW type, they were not observed experimentally. With respect to the most stable non-ZW heterochiral and homochiral dimer structures,
R,
S-HSerAsn
+ Type II, #3 has a drastically different IR pattern compared to that of
S,
S-HSerAsn
+ Type I, #2. This prediction appears to
contradict the previous statement, that experimentally only minor differences are present between the homo- and heterochiral IRMPD spectra (
Figure 6). Furthermore, the observed heterochiral IRMPD spectra of HSerAsn
+ can be accounted for mainly by
R,
S-HSerAsn
+ Type I structures, with essentially no contribution from the
R,
S-HSerAsn
+ Type II, #3 isomer, the most stable non-ZW form of
R,
S-HSerAsn
+ predicted. While the predicted energy ordering or gaps may not be totally trustworthy, the level of theory used here has generally captured the energy ordering of similar neutral species quite well, as demonstrated by many examples reported by the rotational spectroscopic community [
53,
54]. On the other hand, kinetically trapped neutral species in a jet expansion [
11] and in electrosprayed ions [
55] have been reported before. To explain the observation discussed above, we also examine if the amino acid dimers are mainly formed in solution or in the gas phase during the electrospray processes and the influence of the relative stabilities (i.e., abundances) of the monomeric subunits.
In
Table 1, we list the monomeric composition of the most stable isomers of each type for HSerAsn
+ and HValAsn
+, while the related results of all low energy binary isomers are provided in
Table S5. In a previous study, Zhu et al. [
56] estimated the degree of self-aggregation of Ser in water with respect to the concentration. Eight different concentrations, ranging from 0.1 M to saturation were studied, and no severe self-aggregation was observed in any of them. The concentration of our sample solution is ~3 mM, much lower than the concentrations used in the previous study, indicating that the formation of amino acid dimers in the millimolar solution is likely to be negligibly small. HSerAsn
+ and HValAsn
+ are probably formed mainly during the electrospray process. While amino acids exist mainly as zwitterions in a pH (near) neutral aqueous solution, an isolated amino acid exists dominantly in a non-ZW form in the gas phase. This is the case even for the most basic amino acid, arginine [
57]. The poor stability of the zwitterionic form in the gas phase provides an explanation for the non-observation of any binary species with zwitterionic subunits, irrespective of their predicted relative free energies.
The homo- and heterochiral HSerAsn
+ and HValAsn
+ which have been assigned in the experimental IRMPD spectra are shaded in
Table 1. It is interesting to note that all of them contain the most stable monomeric protonated species: HS1
+, HV1
+ and HA
+. We note that HS1
+ identified here is also the most stable isomer reported in the previous IRMPD studies by Wu et al. [
18] and Sunahori et al. [
24]. HV1
+ corresponds to the most stable conformation reported for the protonated monomer of valine methyl esters [
58]. Two similar HA
+ configurations are utilized in the dimers and correspond to the two most stable protonated Asn isomers reported by Heaton and co-workers [
59] and by Heger et al. [
60], with their structures differing slightly depending on which carbonyl O lone pair is used in the intramolecular H-bond with the NH
3+ group.
The connection to the stability of the neutral monomeric species is less clear. For example, the observed Type I
S,
S-HSerAsn
+ is made of HS1
+/A1, where A1 (
cis-COOH) has a very similar configuration to Ic, a higher energy isomer, reported in a previous jet microwave spectroscopic study [
32]. The non-observed Type III
S,
S-HSerAsn
+, on the other hand, is made of HS1
+/A2, where A2 (
trans-COOH) takes on a structure somewhere between II
a, the only one observed experimentally in a jet, and II
b, a much higher energy isomer [
32]. The observed Type III structure of HSerAsn
+ is made of S1/A
+, where S1 has a structure somewhere between I
b and I′
b, two higher isomers of Ser [
30]. These observations are not too surprising because in a protonated dimer, the neutral subunit often opens up some of its intramolecular hydrogen bonds to accommodate a strong intermolecular interaction with its protonated counterpart. It is noted that structural interconversions, for example, the proton migration which is discussed in
Section 2.5, may complicate the discussion of the formation of gas-phase ions, although one would expect such processes to lead to more stable species. Overall, the abundances of the monomeric ZW and protonated subunits in the gas phase seems to play an important role in which dimer can be observed experimentally, rather than just the relative thermodynamic stability of the dimers.
To explore the possibility that chirality recognition effects may be detected in some other frequency regions, the predicted IR spectra in the 0–2650 cm
−1 region of all the
assigned structures, namely Type I and Type III structures of HSerAsn
+ and Type I structures of HValAsn
+ are depicted in
Figure S5,
Supplementary Material. The zoom-in spectra in the 1000–1900 cm
−1 region are also shown. The 1600–1700 cm
−1 region offers potentially the most noticeably different IR band features which are associated with the NH
x scissoring of the protonated NH
3+ and sidechain NH
2 functional groups, as well as the NH
3 umbrella bending motion.
Very recently, Andersson et al. reported minor differences in the IRMPD spectra of the homo- and heterochiral proton-bound Asn dimer [
61], similar to what we observe in terms of the chirality effects in the IRMPD spectra of the four species discussed. Based on their experiment and also a related theoretical study [
37], the authors suggested that to observe chiral differences within the mid-IR region, a sidechain must be involved in the intermolecular interactions. This hypothesis also appears to apply in the simulated IR spectra of several types of the current species. For example, Type II
S,
S- and
R,
S-HSerAsn
+ isomers have
both sidechains of Ser and Asn involved in intermolecular interactions, and noticeable chiral effects, i.e., differences in the homo versus heterochiral IR spectra, are predicted. On the other hand, Type III structures have the least sidechain involvement in the intermolecular interactions and their homo- and heterochiral IR spectra are more similar, showing almost no chiral effects. For the protonated Asn dimers, the authors also suggested that dimers with limited interactions with the sidechain are energetically favored. This does not appear to apply to the current systems since the most favored binary species (based on the theoretical and experimental results) are Type I structures which have more or similar sidechain involvement as compared to the other Types of structures.
It is interesting to point out that the ZW types of HSerAsn
+ and HValAsn
+ are predicted to have a slight homochiral preference (see
Figure 3 and
Figure 4), consistent with the homochiral preference trend reported in Ref. [
29], where in a racemic mixture of Ser or Val with an excess of
R-Asn,
R-Ser or
R-Val preferentially co-crystallized, respectively. Since none of these ZW types were observed in the current experimental study, to appreciate the role that chirality plays in the energy ordering, we carried out noncovalent interaction (NCI) analyses [
62] and quantum theory of atoms in molecules (QTAIM) [
63] analyses of the experimentally observed Type I binary homo- and heterochiral HSerAsn
+ species. The NCI analyses are depicted in
Figure 8. As one can see in
Figure 8, the intermolecular hydrogen bonds are from the NH
3+ group of Ser to the NH
2 and the O of the carbonyl group of Asn in both homo- and heterochiral dimers, whereas Ser has its carboxyl and the hydroxyl groups pointing away from Asn. The NCI plots also show other intermolecular interactions besides the two hydrogen bonds mentioned above, more for
S,
S- than
R,
S-HSerAsn
+. To quantify the strength of the main hydrogen bonds, we also carried out a QTAIM analysis and used the recently derived equation for charged complexes to estimate the associated intermolecular interaction bond energies [
64]. The bond energies of the intermolecular N–H
+⋯O interactions for the homo- and heterochiral HSerAsn
+ are 44.7 kJ mol
−1 and 41.7 kJ mol
−1, respectively, very similar in strength. In contrast, the bond energies of the N–H
+⋯N intermolecular hydrogen bonds for the homo- and heterochiral HSerAsn
+ are 68.2 and 57.5 kJ mol
−1, respectively, ~11 kJ mol
−1 smaller for
R,
S-HSerAsn
+ than for
S,
S-HserAsn
+. It appears that constrained by its chirality, it is more difficult for
R,
S-HserAsn
+ to optimize its intermolecular interactions while it sustains/minimizes the attractive/repulsive intramolecular interactions simultaneously, than
S,
S-HserAsn
+. Overall, a noticeable chirality recognition energy gap of 8.3 kJ mol
−1 (ZPE corrected energy) and 2.3 kJ mol
−1 (free energy) respectively, is present for
R,
S- versus
S,
S-HserAsn
+, with the latter being more stable.
In the case of Type I
S,
S- and
R,
S-HvalAsn
+, the NCI plots are provided in
Figure S6,
Supplementary Material. The corresponding QTAIM intermolecular N–H
+⋯O bond energies are 42.5 and 35.3 kJ mol
−1 for the Type I
S,
S- and
R,
S-HValAsn
+, respectively. For the N–H
+⋯N bond, these values are 70.3 and 65.3 kJ mol
−1 for the Type I
S,
S- and
R,
S-HValAsn
+, respectively. Overall, the chirality recognition energy is about only 1.7 kJ mL
−1 (ZPE corrected energy) and 0.7 kJ mol
−1 (free energy) in favour of the homochiral dimer. A plausible explanation is that Val has a nonpolar sidechain which is less involved in noncovalent interactions, leading to less influence on chirality recognition. As a result, the chirality recognition energy of this system is relatively small by comparison to that of HSerAsn
+.
In comparison to the typical chirality recognition energies encountered in the neutral homo- and heterochiral dimers, for example in those containing transient chiral subunits investigated using jet-cooled FTMW spectroscopy [
65,
66], the values predicted for the current series of homo- and heterochiral dimers are much larger. Certainly, the very high spectral resolution associated with jet-cooled FTMW spectroscopy provides a significant advantage in resolving possible conformers, over the current IRMPD spectroscopy. If one can lower the experimental temperature to, for example 100 K, one would reduce the number of populating isomers and obtain a less crowded spectrum. At 100 K, one would expect only Type I, #2 for
S,
S-HSerAsn
+, and Type I, #7 for
R,
S-HSerAsn
+ based on both the thermodynamic and kinetic controlled processes discussed, leading to more obvious differences between the homo- and heterochiral IR spectra (
Figure 6). Indeed, a recent low temperature study of protonated glutamic acid dimers demonstrated that a cryogenic temperature approach could offer more clarity on chirality recognition spectral signatures [
67].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}