
Physiology-Based Pharmacokinetic Study on 18β-Glycyrrhetic Acid Mono-Glucuronide (GAMG) Prior to Glycyrrhizin in Rats

 and
and
Abstract
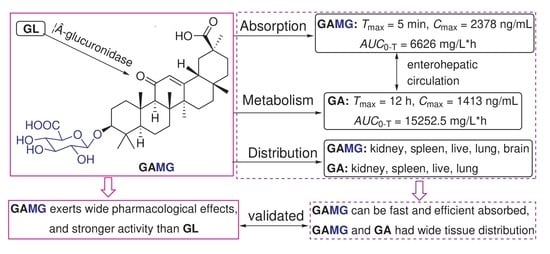
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HPLC-MS/MS Conditions
2.3. Preparation of Calibration Standard QC Samples
2.4. Plasma and Tissue Sample Processing Methods
2.5. Methodology Validation
2.5.1. Specificity
2.5.2. Linearity and Lower Limits of Quantification (LLOQ)
2.5.3. Accuracy and Precision
2.5.4. Recovery and Matrix Effect
2.5.5. Stability
2.5.6. Dilution Reliability
2.6. Animal Experiments and Sampling
2.7. Statistical Analysis
3. Results and Discussion
3.1. Chromatographic and Mass Spectrometric Condition Optimization
3.2. Method Validation
3.2.1. Specificity
3.2.2. Calibration Curve and Linearity
3.2.3. Accuracy and Precision
3.2.4. Recovery and Matrix Effect
3.2.5. Stability
3.2.6. Dilution Integrity
3.3. Pharmacokinetic Studies
3.3.1. Dose Correlation and Gender Differences
3.3.2. Comparison of Pharmacokinetics between GAMG Group and GL Group
3.4. Tissue Distribution Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Hong, S.H.; Kim, B.T.; Bae, E.A.; Park, H.Y.; Han, M.J. Biotransformation of glycyrrhizin by human intestinal bacteria and its relation to biological activities. Arch. Pharm. Res. 2000, 23, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Feng, X.; Jia, J.; Chen, X.; Jiang, T.; Rasool, A.; Lv, B.; Qu, L.; Li, C. A Novel β-Glucuronidase from Talaromyces pinophilus Li-93 Precisely Hydrolyzes Glycyrrhizin into Glycyrrhetinic Acid 3-O-Mono-β-D-Glucuronide. Appl. Environ. Microbiol. 2018, 84, e00755-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.J.; Yang, Y.A.; Xu, H.; Shi, J.B.; Liu, X.H. Synthesis and discovery of 18α-GAMG as anticancer agent in vitro and in vivo via down expression of protein p65. Sci. Rep. 2014, 4, 7106. [Google Scholar] [CrossRef]
- Li, B.; Yang, Y.; Chen, L.; Chen, S.; Zhang, J.; Tang, W. 18alpha-Glycyrrhetinic acid monoglucuronide as an anti-inflammatory agent through suppression of the NF-kappaB and MAPK signaling pathway. Medchemcomm 2017, 8, 1498–1504. [Google Scholar] [CrossRef]
- Zhang, X.L.; Li, B.; Zhang, X.; Zhu, J.; **e, Y.; Shen, T.; Tang, W.; Zhang, J. 18β-Glycyrrhetinic acid monoglucuronide (GAMG) alleviates single-walled carbon nanotubes (SWCNT)-induced lung inflammation and fibrosis in mice through PI3K/AKT/NF-κB signaling pathway. Ecotoxicol. Environ. Saf. 2022, 242, 113858. [Google Scholar] [CrossRef]
- Park, H.Y.; Park, S.H.; Yoon, H.K.; Han, M.J.; Kim, D.H. Anti-allergic activity of 18β-glycyrrhetinic acid-3-O-β-D-glucuronide. Arch. Pharm. Res. 2004, 27, 57–60. [Google Scholar] [CrossRef]
- Guo, L.; Katiyo, W.; Lu, L.; Zhang, X.; Wang, M.; Yan, J.; Ma, X.; Yang, R.; Zou, L.; Zhao, W. Glycyrrhetic Acid 3-O-Mono-β-d-glucuronide (GAMG): An Innovative High-Potency Sweetener with Improved Biological Activities. Compr. Rev. Food Sci. Food Saf. 2018, 17, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wei, Y.; Guo, X.; Qi, P.; Zhu, H.; Tang, W. Glycyrrhetic acid monoglucuronide: Sweetness concentration-response and molecular mechanism as a naturally high-potency sweetener. Food Sci. Biotechnol. 2019, 28, 1187–1193. [Google Scholar] [CrossRef]
- Kawakami, J.; Yamamura, Y.; Santa, T.; Kotaki, H.; Uchino, K.; Sawada, Y.; Iga, T. Kinetic analysis of glycyrrhetic acid, an active metabolite of glycyrrhizin, in rats: Role of enterohepatic circulation. J. Pharm. Sci. 1993, 82, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ploeger, B.A.; Meulenbelt, J.; DeJongh, J. Physiologically based pharmacokinetic modeling of glycyrrhizic acid, a compound subject to presystemic metabolism and enterohepatic cycling. Toxicol. Appl. Pharmacol. 2000, 162, 177–188. [Google Scholar] [CrossRef]
- Feng, X.; Ding, L.; Qiu, F. Potential drug interactions associated with glycyrrhizin and glycyrrhetinic acid. Drug Metab. Rev. 2015, 47, 229–238. [Google Scholar] [CrossRef]
- Li, F.Y.; **e, H.; Weng, L.; Wang, H.; Cao, L.J.; Hao, H.P.; Wang, G.J. Effects of diammonium glycyrrhizinate on hepatic and intestinal UDP-Glucuronosyltransferases in rats: Implication in herb-drug interactions. Chin. J. Nat. Med. 2016, 14, 534–540. [Google Scholar] [CrossRef]
- Dong, J.; Olaleye, O.E.; Jiang, R.; Li, J.; Lu, C.; Du, F.; Xu, F.; Yang, J.; Wang, F.; Jia, W.; et al. Glycyrrhizin has a high likelihood to be a victim of drug-drug interactions mediated by hepatic organic anion-transporting polypeptide 1B1/1B3. Br. J. Pharmacol. 2018, 175, 3486–3503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zou, C.; Qin, M.; Li, Y.; Huang, J. A simple method for evaluation pharmacokinetics of glycyrrhetinic acid and potential drug-drug interaction between herbal ingredients. Sci. Rep. 2019, 9, 11308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ren, S.; Liu, X.; Liu, X.; Guo, F.; Sun, W.; Feng, X.; Li, C. Mining of UDP-glucosyltrfansferases in licorice for controllable glycosylation of pentacyclic triterpenoids. Biotechnol. Bioeng. 2020, 117, 3651–3663. [Google Scholar] [CrossRef]
- Nomura, Y.; Seki, H.; Suzuki, T.; Ohyama, K.; Mizutani, M.; Kaku, T.; Tamura, K.; Ono, E.; Horikawa, M.; Sudo, H.; et al. Functional specialization of UDP-glycosyltransferase 73P12 in licorice to produce a sweet triterpenoid saponin, glycyrrhizin. Plant J. 2019, 99, 1127–1143. [Google Scholar] [CrossRef] [Green Version]
- Fan, R. Separation of Glycyrrhizic Acid and Its Derivants from Hydrolyzation in Subcritical Water by Macroporous Resin. Molecules 2020, 25, 4305. [Google Scholar] [CrossRef]
- Jiang, T.; Hou, Y.; Zhang, T.; Feng, X.; Li, C. Construction of a CaHPO4-PGUS1 hybrid nanoflower through protein-inorganic self-assembly, and its application in glycyrrhetinic acid 3-O-mono-β-d-glucuronide preparation. Front. Chem. Sci. Eng. 2019, 13, 554–562. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, B.; **ao, Y.; Yang, H.; Wang, Y.; Du, L.; Zhu, D. Purification and characterization of a novel β-glucuronidase precisely converts glycyrrhizin to glycyrrhetinic acid 3-O-mono-β-D-glucuronide from plant endophytic Chaetomium globosum DX-THS3. Int. J. Biol. Macromol. 2020, 159, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.H.; Zhang, L.L.; Li, T.; Lu, J.H.; Ma, D.L.; Leung, C.H.; Chen, X.P.; Jiang, H.L.; Wang, Y.T.; Lu, J.J. Glycyrrhetinic acid induces cytoprotective autophagy via the inositol-requiring enzyme 1α-c-Jun N-terminal kinase cascade in non-small-cell lung cancer cells. Oncotarget 2015, 6, 43911–43926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khateeb, E.; Burkhill, S.; Murby, S.; Amirat, H.; Rostami-Hodjegan, A.; Ahmad, A. Physiological-based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20-years; in-depth analysis of applications, organizations, and platforms. Biopharm. Drug Dispos. 2021, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Dallmann, A.; Frechen, S.; Coboeken, K.; Niederalt, C.; Wendl, T.; Block, M.; Meyer, M.; Eissing, T.; Burghaus, R.; et al. Predictive Performance of Physiology-Based Pharmacokinetic Dose Estimates for Pediatric Trials: Evaluation with 10 Bayer Small-Molecule Compounds in Children. J. Clin. Pharmacol. 2021, 61 (Suppl. S1), S70–S82. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tsukahara, M.; Akasaka, Y.; Inoue, H. A highly sensitive LC–MS/MS method for simultaneous determination of glycyrrhizin and its active metabolite glycyrrhetinic acid: Application to a human pharmacokinetic study after oral administration. Biomed. Chromatogr. 2017, 31, 4032–4037. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosamples | Linear Range (ng/mL) | Calibration Curves | Correlation Coeffcient (r) | LLOQs (ng/mL) | RSD of LLOQs (n = 6, %) | RE of LLOQs (n = 6, %) |
|---|---|---|---|---|---|---|
| Plasma | 2–300 | Y = 0.3823X − 0.0239 | 0.999 | 2 | 3.35 | 7.50 |
| Heart | 10–1000 | Y = 1.4309X + 0.4599 | 0.996 | 10 | 3.96 | 11.00 |
| Liver | 10–2000 | Y = 1.4784X + 0.3639 | 0.998 | 10 | 5.85 | 7.23 |
| Spleen | 10–2000 | Y = 1.0012X + 0.5079 | 0.999 | 10 | 9.11 | 9.40 |
| Lung | 10–1000 | Y = 0.9201X + 0.9474 | 0.997 | 10 | 8.30 | 12.40 |
| Kidney | 10–2000 | Y = 0.8114X + 0.4544 | 0.999 | 10 | 8.26 | 12.76 |
| Brain | 10–2000 | Y = 0.9922X + 0.0052 | 0.998 | 10 | 2.70 | 5.45 |
| Small Intestine | 10–2000 | Y = 0.3612X + 0.0062 | 0.999 | 10 | 7.48 | 12.92 |
| Biosamples | Linear Range (ng/mL) | Calibration Curves | Correlation Coeffcient (r) | LLOQs (ng/mL) | RSD of LLOQs (n = 6, %) | RE of LLOQs (n = 6, %) |
|---|---|---|---|---|---|---|
| Plasma | 0.5–150 | Y = 0.9184X + 0.1610 | 0.998 | 0.5 | 14.74 | 6.96 |
| Heart | 5–500 | Y = 0.4259X − 0.0296 | 0.998 | 5 | 7.32 | 13.11 |
| Liver | 5–1000 | Y = 0.4925X − 0.0781 | 0.996 | 5 | 5.82 | 11.99 |
| Spleen | 1–200 | Y = 0.4261X − 0.0052 | 0.998 | 1 | 4.09 | 8.90 |
| Lung | 5–500 | Y = 0.2573X + 0.0066 | 0.996 | 5 | 5.34 | 13.82 |
| Kidney | 5–200 | Y = 0.2164X + 0.0263 | 0.996 | 5 | 12.20 | 10.28 |
| Brain | 5–200 | Y = 0.2201X + 0.0267 | 0.997 | 5 | 4.00 | 12.51 |
| Small Intestine | 1–200 | Y = 0.2292X + 0.0723 | 0.998 | 1 | 8.20 | 11.58 |
| Analyte | Theoretical Concentration (ng/mL) | Intraday (n = 6) | Interday (n = 6) | ||||
|---|---|---|---|---|---|---|---|
| Mean Measure Concentration | RSD (%) | RE (%) | Mean Measure Concentration | RSD (%) | RE (%) | ||
| GAMG | 3 | 3.31 | 14.20 | 10.33 | 3.08 | 12.99 | 2.67 |
| 40 | 41.26 | 1.94 | 3.15 | 42.70 | 5.36 | 6.75 | |
| 250 | 253.74 | 4.64 | 1.5 | 258.99 | 4.31 | 3.60 | |
| GA | 1.5 | 1.71 | 13.28 | 14.00 | 1.67 | 11.24 | 11.33 |
| 20 | 22.95 | 10.54 | 14.75 | 22.02 | 12.85 | 10.10 | |
| 120 | 121.33 | 6.33 | 1.11 | 113.98 | 8.86 | −5.02 | |
| Analyte | Theoretical Concentration (ng/mL) | Sample Extraction Recovery Rate (%, Mean ± SD) | Internal Standard Extraction Recovery Rate (%, Mean ± SD) | Sample Extraction Recovery Rate (%, RSD) | Internal Standard Extraction Recovery Rate (%, RSD) | MFSample% (%, Mean ± SD) | MFIS% (%, Mean ± SD) | Matrix Factor Normalized by Internal Standard (%, CV) |
|---|---|---|---|---|---|---|---|---|
| GAMG | 3 | 68.94 ± 8.76 | 97.01 ± 9.05 | 12.71 | 9.05 | 78.12 ± 8.44 | 75.43 ± 7.43 | 11.77 |
| 40 | 73.84 ± 6.92 | 70.34 ± 7.14 | 9.37 | 10.15 | 126.14 ± 7.55 | 113.63 ± 15.48 | 12.91 | |
| 250 | 109.94 ± 8.33 | 97.62 ± 9.96 | 7.58 | 9.96 | 76.46 ± 7.12 | 97.40 ± 12.25 | 7.20 | |
| GA | 1.5 | 104.96 ± 11.41 | 102.12 ± 12.09 | 10.87 | 11.84 | 67.30 ± 6.47 | 71.59 ± 8.61 | 6.88 |
| 20 | 93.07 ± 8.41 | 70.27 ± 6.13 | 9.04 | 11.94 | 89.36 ± 5.48 | 113.06 ± 9.07 | 8.89 | |
| 120 | 119.61 ± 10.49 | 97.53 ± 8.82 | 8.77 | 9.04 | 83.08 ± 5.56 | 97.19 ± 9.94 | 13.60 |
| Analyte | Theoretical Concentration (ng/mL) | Short-Term Room Temperature Stability at 25 °C, 4 h (%, Mean ± SD) | Short-Term Room Temperature Stability at 25 °C, 4 h (%, RE) | Long-Term Frozen Storage Stability at −40 °C (%, Mean ± SD) | Long-Term Frozen Storage Stability at −40 °C (%, RE) | Repeated Freeze-Thaw Stability (%, Mean ± SD) | Repeated Freeze-thaw Stability (%, RE) |
|---|---|---|---|---|---|---|---|
| GAMG | 3 | 2.87 ± 0.12 | −4.33 | 2.78 ± 0.06 | −7.33 | 2.73 ± 0.03 | −9.00 |
| 40 | 45.45 ± 0.10 | 13.62 | 35.12 ± 0.58 | 1.66 | 38.07 ± 0.88 | −4.82 | |
| 250 | 251.81 ± 5.15 | 0.72 | 212.88 ± 3.84 | 14.85 | 228.37 ± 9.46 | −8.65 | |
| GA | 1.5 | 2.43 ± 0.07 | −2.8 | 2.37 ± 0.14 | −5.20 | 2.41 ± 0.34 | −3.60 |
| 20 | 21.55 ± 0.31 | 7.55 | 17.63 ± 0.33 | −11.85 | 18.45 ± 0.28 | −7.75 | |
| 120 | 121.23 ± 1.91 | 1.03 | 107.62 ± 3.35 | 3.11 | 107.62 ± 3.35 | −10.32 |
| Pharmacokinetic Parameters | p Value | ||
|---|---|---|---|
| Low Dose Group | Medium Dose Group | High Dose Group | |
| Cmax (ng/mL) | 0.874 | 0.710 | 0.941 |
| Tmax (h) | 1.00 | 1.00 | 1.00 |
| T1/2 (h) | 0.605 | 0.733 | 0.423 |
| AUC0-t (ng/mL*h) | 0.244 | 0.058 | 0.960 |
| MRT0-t (h) | 0.369 | 0.474 | 0.831 |
| Vd (mL/kg) | 0.946 | 0.377 | 0.348 |
| CL (mL/h/kg) | 0.232 | 0.083 | 0.667 |
| Pharmacokinetic Parameters | p Value | ||
|---|---|---|---|
| Low Dose Group | Medium Dose Group | High Dose Group | |
| Cmax (ng/mL) | 0.688 | 0.229 | 0.412 |
| Tmax (h) | 0.801 | 0.134 | 0.391 |
| T1/2 (h) | 0.570 | 0.777 | 0.124 |
| AUC0-t (ng/mL*h) | 0.244 | 0.157 | 0.682 |
| MRT0-t (h) | 0.499 | 0.714 | 0.508 |
| Vd (mL/kg) | 0.838 | 0.361 | 0.302 |
| CL (mL/h/kg) | 0.638 | 0.163 | 0.493 |
| PK Parameters | GL Group (n = 6, Mean ± SD) | GAMG Group (n = 6, Mean ± SD) |
|---|---|---|
| Cmax (ng/mL) | 346.03 ± 145.13 | 2377.57 ± 547.40 |
| Tmax (h) | 2.00 ± 0.00 | 0.083 ± 0.00 |
| T1/2 (h) | 8.18 ± 2.48 | 15.73 ± 7.26 |
| AUC0-T (mg/L*h) | 459.32 ± 80.81 | 6625.54 ± 1680.70 |
| MRT0-T (h) | 17.54 ± 2.81 | 11.22 ± 2.58 |
| Vd (mL/kg) | 133.15 ± 41.06 | 99.25 ± 56.43 |
| CL (mL/h/kg) | 11.30 ± 1.93 | 4.85 ± 1.59 |
| PK Parameters | GL Group (n = 6, Mean ± SD) | GAMG Group (n = 6, Mean ± SD) |
|---|---|---|
| Cmax (ng/mL) | 747.08 ± 236.85 | 1412.58 ± 80.83 |
| Tmax (h) | 13.67 ± 0.82 | 12.33 ± 0.82 |
| T1/2 (h) | 7.54 ± 2.86 | 8.48 ± 5.00 |
| AUC0-T (mg/L*h) | 11,598.49 ± 4496.08 | 15,252.54 ± 4661.22 |
| MRT0-T (h) | 18.14 ± 2.35 | 15.99 ± 1.07 |
| Vd (mL/kg) | 32.06 ± 15.70 | 32.26 ± 36.29 |
| CL (mL/h/kg) | 3.00 ± 1.38 | 2.20 ± 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Zuo, J.; Yang, J.-G.; Wu, C.; Yang, Y.; Tang, W.; Zhu, L. Physiology-Based Pharmacokinetic Study on 18β-Glycyrrhetic Acid Mono-Glucuronide (GAMG) Prior to Glycyrrhizin in Rats. Molecules 2022, 27, 4657. https://doi.org/10.3390/molecules27144657
Cao M, Zuo J, Yang J-G, Wu C, Yang Y, Tang W, Zhu L. Physiology-Based Pharmacokinetic Study on 18β-Glycyrrhetic Acid Mono-Glucuronide (GAMG) Prior to Glycyrrhizin in Rats. Molecules. 2022; 27(14):4657. https://doi.org/10.3390/molecules27144657
Chicago/Turabian StyleCao, Mengxin, Jiawei Zuo, Jian-Guo Yang, Chenyao Wu, Yongan Yang, Wenjian Tang, and Lili Zhu. 2022. "Physiology-Based Pharmacokinetic Study on 18β-Glycyrrhetic Acid Mono-Glucuronide (GAMG) Prior to Glycyrrhizin in Rats" Molecules 27, no. 14: 4657. https://doi.org/10.3390/molecules27144657