
Anti-Inflammatory Activity of 4-(4-(Heptyloxy)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one via Repression of MAPK/NF-κB Signaling Pathways in β-Amyloid-Induced Alzheimer’s Disease Models

Abstract
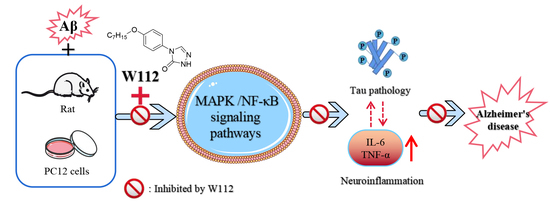
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effects of W112 on Cell Survival in Aβ25–35-Induced PC12 Cells
2.2. Effects of W112 on Spatial Learning and Memory Abilities in Rats
2.3. Effects of W112 on Aβ25–35-Induced Tau Hyperphosphorylation
2.4. Effects of W112 on the Aβ25–35-Induced Neuroinflammation
2.5. Effects of W112 on Inhibition of NF-κB Signaling Pathway
2.6. Effects of W112 on MAPK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Preparation of Aβ25–35
4.2. Cell Culture and Viability Assays
4.3. Animal and Treatments
4.4. MWM Test
4.5. Immunohistochemistry
4.6. Western Blotting Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef] [PubMed]
- Rank, K.B.; Pauley, A.M.; Bhattacharya, K.; Wang, Z.; Evans, D.B.; Fleck, T.J.; Johnston, J.A.; Sharma, S.K. Direct interaction of soluble human recombinant tau protein with Abeta 1-42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett. 2002, 514, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, B.; Mehra, V.; Kumar, V. Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1,2,3-triazoles. Eur. J. Med. Chem. 2021, 212, 113069. [Google Scholar] [CrossRef]
- Liu, X.J.; Zhang, H.J.; Quan, Z.S. Synthesis and evaluation of the anticonvulsant activities of 2,3-dihydrophthalazine-1,4-dione derivatives. Med. Chem. Res. 2017, 26, 1935–1946. [Google Scholar] [CrossRef]
- Zhang, G.R.; Ren, Y.; Yin, X.M.; Quan, Z.S. Synthesis and Evaluation of the Anticonvulsant Activities of New 5-substitued-[1,2,4]triazolo [4,3-a]quinoxalin-4(5H)-one Derivatives. Lett. Drug Des. Discov. 2018, 15, 406–413. [Google Scholar] [CrossRef]
- Fang, Y.Q.; Sun, C.L.; Liu, D.C.; Wang, S.B.; Quan, Z.S. Synthesis and Anticonvulsant Activity Evaluation of 3-alkoxy-4-(4-(hexyloxy/heptyloxy)phenyl)-4H-1,2,4-triazole. Iran. J. Pharm. Res. 2015, 14, 77–87. [Google Scholar] [PubMed]
- Özil, M.; Balaydın, H.T.; Şentürk, M. Synthesis of 5-methyl-2,4-dihydro-3H-1,2,4-triazole-3-one’s aryl Schiff base derivatives and investigation of carbonic anhydrase and cholinesterase (AChE, BuChE) inhibitory properties. Bioorg. Chem. 2019, 86, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Zhang, J.; Rodrigues, M.C.; Ding, D.J.; Longo, J.P.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.S. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Mann, S.; Kaur, A.; Priyadarshi, N.; Goyal, B.; Singhal, N.K.; Goyal, D. Multi-target-directed triazole derivatives as promising agents for the treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 87, 572–584. [Google Scholar] [CrossRef]
- Fronza, M.G.; Baldinotti, R.; Sacramento, M.; Gutierres, J.; Carvalho, F.B.; Fernandes, M.D.C.; Sousa, F.S.S.; Seixas, F.K.; Collares, T.; Alves, D.; et al. Effect of QTC-4-MeOBnE Treatment on Memory, Neurodegeneration, and Neurogenesis in a Streptozotocin-Induced Mouse Model of Alzheimer’s Disease. ACS. Chem. Neurosci. 2021, 12, 109–122. [Google Scholar] [CrossRef]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimer’s Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Sharma, N.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Felemban, S.G.; Alsubayiel, A.M.; et al. “Aducanumab” making a comeback in Alzheimer’s disease: An old wine in a new bottle. Biomed. Pharmacother. 2022, 148, 112746. [Google Scholar] [CrossRef]
- Gitto, R.; Vittorio, S.; Bucolo, F.; Peña-Díaz, S.; Siracusa, R.; Cuzzocrea, S.; Ventura, S.; Di Paola, R.; De Luca, L. Discovery of Neuroprotective Agents Based on a 5-(4-Pyridinyl)-1,2,4-triazole Scaffold. ACS Chem. Neurosci. 2022, 13, 581–586. [Google Scholar] [CrossRef]
- Wu, J.; Hou, Z.; Wang, Y.; Chen, L.; Lian, C.; Meng, Q.; Zhang, C.; Li, X.; Huang, L.; Yu, H. Discovery of 7-alkyloxy-[1,2,4] triazolo [1,5-a] pyrimidine derivatives as selective positive modulators of GABAA1 and GABAA4 receptors with potent antiepileptic activity. Bioorg. Chem. 2022, 119, 105565. [Google Scholar] [CrossRef]
- Wang, M.; Fang, L.; Liu, T.; Chen, X.; Zheng, Y.; Zhang, Y.; Chen, S.; Li, Z. Discovery of 7-O-1, 2, 3-triazole hesperetin derivatives as multi-target-directed ligands against Alzheimer’s disease. Chem. Biol. Interact. 2021, 342, 109489. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millucci, L.; Raggiaschi, R.; Franceschini, D.; Terstappen, G.; Santucci, A. Rapid aggregation and assembly in aqueous solution of A beta (25–35) peptide. J. Biosci. 2009, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Perez, E.J.; Muñoz, B.; Bascuñan, D.A.; Peters, C.; Riffo-Lepe, N.O.; Espinoza, M.P.; Morgan, P.J.; Filippi, C.; Bourboulou, R.; Sengupta, U.; et al. Synaptic dysregulation and hyperexcitability induced by intracellular amyloid beta oligomers. Aging Cell 2021, 20, e13455. [Google Scholar] [CrossRef]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimer′s Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Horie, K.; Barthélemy, N.R.; Sato, C.; Bateman, R.J. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain 2021, 144, 515–527. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Stimmell, A.C.; Xu, Z.; Moseley, S.C.; Benthem, S.D.; Fernandez, D.M.; Dang, J.V.; Santos-Molina, L.F.; Anzalone, R.A.; Garcia-Barbon, C.L.; Rodriguez, S.; et al. Tau Pathology Profile Across a Parietal-Hippocampal Brain Network Is Associated with Spatial Reorientation Learning and Memory Performance in the 3xTg-AD Mouse. Front. Aging 2021, 2, 655015. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef] [Green Version]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer′s. Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhou, R.; Tong, Y.; Chen, P.; Shen, Y.; Miao, S.; Liu, X. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol. Dis. 2020, 140, 104814. [Google Scholar] [CrossRef]
- Laurent, C.; Dorothée, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Légeron, F.P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Yokokura, M.; Obi, T.; Bunai, T.; Yoshikawa, E.; Ando, I.; Shimada, H.; Suhara, T.; Higuchi, M.; Ouchi, Y. In vivo direct relation of tau pathology with neuroinflammation in early Alzheimer’s disease. J. Neurol. 2019, 266, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, S.W.; Huang, Z.Z.; Shan, F.B.; Li, P.; Ning, Y.L.; Ye, S.Y.; Zhao, Z.A.; Du, H.; **ong, R.P.; et al. NLRP3 Inflammasome-Dependent Increases in High Mobility Group Box 1 Involved in the Cognitive Dysfunction Caused by Tau-Overexpression. Front. Aging Neurosci. 2021, 13, 721474. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- **e, L.; Zhang, N.; Zhang, Q.; Li, C.; Sandhu, A.F.; Iii, G.W.; Lin, S.; Lv, P.; Liu, Y.; Wu, Q.; et al. Inflammatory factors and amyloid β-induced microglial polarization promote inflammatory crosstalk with astrocytes. Aging 2020, 12, 22538–22549. [Google Scholar] [CrossRef]
- Sun, J.; Qin, X.; Zhang, X.; Wang, Q.; Zhang, W.; Wang, M. FBXW11 deletion alleviates Alzheimer’s disease by reducing neuroinflammation and amyloid-β plaque formation via repression of ASK1 signaling. Biochem. Biophys. Res. Commun. 2021, 548, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Walsh, D.M.; Rowan, M.J.; Selkoe, D.J.; Anwyl, R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 2004, 24, 3370–3378. [Google Scholar] [CrossRef] [Green Version]
- Schnöder, L.; Gasparoni, G.; Nordström, K.; Schottek, A.; Tomic, I.; Christmann, A.; Schäfer, K.H.; Menger, M.D.; Walter, J.; Fassbender, K.; et al. Neuronal deficiency of p38α-MAPK ameliorates symptoms and pathology of APP or Tau-transgenic Alzheimer’s mouse models. FASEB J. 2020, 34, 9628–9649. [Google Scholar] [CrossRef]
- Ruganzu, J.B.; Peng, X.; He, Y.; Wu, X.; Zheng, Q.; Ding, B.; Lin, C.; Guo, H.; Yang, Z.; Zhang, X.; et al. Downregulation of TREM2 expression exacerbates neuroinflammatory responses through TLR4-mediated MAPK signaling pathway in a transgenic mouse model of Alzheimer’s disease. Mol. Immunol. 2022, 142, 22–36. [Google Scholar] [CrossRef]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, W.; Plaisance, S.; Boone, E.; De Bosscher, K.; Schmitz, M.L.; Fiers, W.; Haegeman, G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 1998, 273, 3285–3290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.J.; Wang, X.Z.; Cao, Q.; Gong, G.H.; Quan, Z.S. Design, synthesis, anti-inflammatory activity, and molecular docking studies of perimidine derivatives containing triazole. Bioorg. Med. Chem. Lett. 2017, 27, 4409–4414. [Google Scholar] [CrossRef] [PubMed]
- Zhi, T.X.; Liu, K.Q.; Cai, K.Y.; Zhao, Y.C.; Li, Z.W.; Wang, X.; He, X.H.; Sun, X.Y. Anti-Lung Cancer Activities of 1,2,3-Triazole Curcumin Derivatives via Regulation of the MAPK/NF-κB/STAT3 Signaling Pathways. ChemMedChem 2022, 17, e202100676. [Google Scholar] [CrossRef] [PubMed]
- An, F.M.; Liu, Z.; Xuan, X.R.; Liu, Q.S.; Wei, C.X. Sanweidoukou decoction, a Chinese herbal formula, ameliorates β-amyloid protein-induced neuronal insult via modulating MAPK/NF-κB signaling pathways: Studies in vivo and in vitro. J. Ethnopharmacol. 2021, 273, 114002. [Google Scholar] [CrossRef] [PubMed]
- Bruszt, N.; Bali, Z.K.; Tadepalli, S.A.; Nagy, L.V.; Hernádi, I. Potentiation of cognitive enhancer effects of Alzheimer’s disease medication memantine by alpha7 nicotinic acetylcholine receptor agonist PHA-543613 in the Morris water maze task. Psychopharmacology 2021, 238, 3273–3281. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, F.; Xuan, X.; Liu, Z.; Bian, M.; Shen, Q.; Quan, Z.; Zhang, G.; Wei, C. Anti-Inflammatory Activity of 4-(4-(Heptyloxy)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one via Repression of MAPK/NF-κB Signaling Pathways in β-Amyloid-Induced Alzheimer’s Disease Models. Molecules 2022, 27, 5035. https://doi.org/10.3390/molecules27155035
An F, Xuan X, Liu Z, Bian M, Shen Q, Quan Z, Zhang G, Wei C. Anti-Inflammatory Activity of 4-(4-(Heptyloxy)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one via Repression of MAPK/NF-κB Signaling Pathways in β-Amyloid-Induced Alzheimer’s Disease Models. Molecules. 2022; 27(15):5035. https://doi.org/10.3390/molecules27155035
Chicago/Turabian StyleAn, Fengmao, **nran Xuan, Zheng Liu, Ming Bian, Qingkun Shen, Zheshan Quan, Guowei Zhang, and Chengxi Wei. 2022. "Anti-Inflammatory Activity of 4-(4-(Heptyloxy)phenyl)-2,4-dihydro-3H-1,2,4-triazol-3-one via Repression of MAPK/NF-κB Signaling Pathways in β-Amyloid-Induced Alzheimer’s Disease Models" Molecules 27, no. 15: 5035. https://doi.org/10.3390/molecules27155035