
Using Triethylborane to Manipulate Reactivity Ratios in Epoxide-Anhydride Copolymerization: Application to the Synthesis of Polyethers with Degradable Ester Functions


Abstract
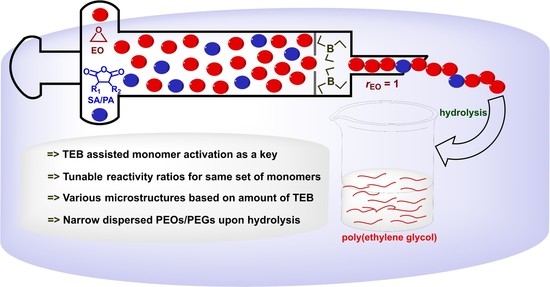
:
1. Introduction
2. Results and Discussion
2.1. Determination of Reactivity Ratio of EO as a Function of the Amount of TEB Used
2.2. Synthesis and Characterization of Degradable PEOs
- (i)
- Reactions with rEO < 1 (entries 1 and 5 in Table 2):
- (ii)
- Reactions with rEO = 0.99 to 1.1 (entries 2, 3, and 6 in Table 2):
- (iii)
- Reactions with rEO > 1 (entries 4 and 7 in Table 2):
3. Materials and Methods
3.1. Materials and Methods
3.2. Representative Procedure for Copolymerization of EO and Anhydride for In Situ Kinetic NMR Experiment
3.3. In Situ Kinetic NMR Experiment Data Processing and Calculation of Reactivity Ratios by Jaacks Model
3.4. Representative Procedure for Copolymerization of EO and Anhydride for Schlenk Tube Experiment
3.5. Representative Procedure for Degradation of PEO Based Poly(ester Ether) Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lutz, J.-F.; Ouchi, M.; Liu, D.R.; Sawamoto, M. Sequence-Controlled Polymers. Science 2013, 341, 1238149. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Lehn, J.-M.; Meijer, E.W.; Matyjaszewski, K. From precision polymers to complex materials and systems. Nat. Rev. Mater. 2016, 1, 16024. [Google Scholar] [CrossRef]
- Grune, E.; Bareuther, J.; Blankenburg, J.; Appold, M.; Shaw, L.; Müller, A.H.E.; Floudas, G.; Hutchings, L.R.; Gallei, M.; Frey, H. Towards bio-based tapered block copolymers: The behaviour of myrcene in the statistical anionic copolymerisation. Polym. Chem. 2019, 10, 1213–1220. [Google Scholar] [CrossRef] [Green Version]
- Blankenburg, J.; Wagner, M.; Frey, H. Well-Defined Multi-Amino-Functional and Stimuli-Responsive Poly(propylene oxide) by Crown Ether Assisted Anionic Ring-Opening Polymerization. Macromolecules 2017, 50, 8885–8893. [Google Scholar] [CrossRef]
- Gleede, T.; Rieger, E.; Blankenburg, J.; Klein, K.; Wurm, F.R. Fast Access to Amphiphilic Multiblock Architectures by the Anionic Copolymerization of Aziridines and Ethylene Oxide. J. Am. Chem. Soc. 2018, 140, 13407–13412. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Zhu, Y.; Dingwall, P.; Paul, S.; Rzepa, H.S.; Buchard, A.; Williams, C.K. Chemoselective Polymerizations from Mixtures of Epoxide, Lactone, Anhydride, and Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 4120–4131. [Google Scholar] [CrossRef] [Green Version]
- Blankenburg, J.; Kersten, E.; Maciol, K.; Wagner, M.; Zarbakhsh, S.; Frey, H. The poly(propylene oxide-co-ethylene oxide) gradient is controlled by the polymerization method: Determination of reactivity ratios by direct comparison of different copolymerization models. Polym. Chem. 2019, 10, 2863–2871. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Inter. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Fowler, R.; Stolnik, S. PEGylated nanomedicines: Recent progress and remaining concerns. Expert Opin. Drug Deliv. 2014, 11, 139–154. [Google Scholar] [CrossRef]
- Lundberg, P.; Lee, B.F.; van den Berg, S.A.; Pressly, E.D.; Lee, A.; Hawker, C.J.; Lynd, N.A. Poly[(ethylene oxide)-co-(methylene ethylene oxide)]: A hydrolytically degradable poly(ethylene oxide) platform. ACS Macro Lett. 2012, 1, 1240–1243. [Google Scholar] [CrossRef] [Green Version]
- Worm, M.; Leibig, D.; Dingels, C.; Frey, H. Cleavable Polyethylene Glycol: 3,4-Epoxy-1-butene as a Comonomer to Establish Degradability at Physiologically Relevant pH. ACS Macro Lett. 2016, 5, 1357–1363. [Google Scholar] [CrossRef]
- Varghese, J.K.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Degradable poly(ethylene oxide) through metal-free copolymerization of ethylene oxide with l-lactide. Polym. Chem. 2019, 10, 3764–3771. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Boopathi, S.K.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Metal-Free Alternating Copolymerization of CO2 with Epoxides: Fulfilling “Green” Synthesis and Activity. J. Am. Chem. Soc. 2016, 138, 11117–11120. [Google Scholar] [CrossRef]
- Chidara, V.K.; Boopathi, S.K.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Triethylborane-Assisted Synthesis of Random and Block Poly(ester-carbonate)s through One-Pot Terpolymerization of Epoxides, CO2, and Cyclic Anhydrides. Macromolecules 2021, 54, 2711–2719. [Google Scholar] [CrossRef]
- Jia, M.; Zhang, D.; Gnanou, Y.; Feng, X. Surfactant-Emulating Amphiphilic Polycarbonates and Other Functional Polycarbonates through Metal-Free Copolymerization of CO2 with Ethylene Oxide. ACS Sustain. Chem. Eng. 2021, 9, 10370–10380. [Google Scholar] [CrossRef]
- Li, H.; He, G.; Chen, Y.; Zhao, J.; Zhang, G. One-Step Approach to Polyester–Polyether Block Copolymers Using Highly Tunable Bicomponent Catalyst. ACS Macro Lett. 2019, 8, 973–978. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, J.; Liu, S.; Zhao, J.; Wang, Y.; Zhang, G. High Efficiency Organic Lewis Pair Catalyst for Ring-Opening Polymerization of Epoxides with Chemoselectivity. Macromolecules 2018, 51, 8286–8297. [Google Scholar] [CrossRef]
- Jaacks, V. A novel method of determination of reactivity ratios in binary and ternary copolymerizations. Makromol. Chem. 1972, 161, 161–172. [Google Scholar] [CrossRef]
- Kummari, A.; Pappuru, S.; Chakraborty, D. Fully alternating and regioselective ring-opening copolymerization of phthalic anhydride with epoxides using highly active metal-free Lewis pairs as a catalyst. Polym. Chem. 2018, 9, 4052–4062. [Google Scholar] [CrossRef]
- Hu, L.-F.; Zhang, C.-J.; Wu, H.-L.; Yang, J.-L.; Liu, B.; Duan, H.-Y.; Zhang, X.-H. Highly Active Organic Lewis Pairs for the Copolymerization of Epoxides with Cyclic Anhydrides: Metal-Free Access to Well-Defined Aliphatic Polyesters. Macromolecules 2018, 51, 3126–3134. [Google Scholar] [CrossRef]
- Ji, H.-Y.; Song, D.-P.; Wang, B.; Pan, L.; Li, Y.-S. Organic Lewis pairs for selective copolymerization of epoxides with anhydrides to access sequence-controlled block copolymers. Green Chem. 2019, 21, 6123–6132. [Google Scholar] [CrossRef]
- DiCiccio, A.M.; Longo, J.M.; Rodriguez-Calero, G.G.; Coates, G.W. Development of Highly Active and Regioselective Catalysts for the Copolymerization of Epoxides with Cyclic Anhydrides: An Unanticipated Effect of Electronic Variation. J. Am. Chem. Soc. 2016, 138, 7107–7113. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Entry | Anhydride (AH) | TBACl:TEB:AH:EO | % Conv. (EO) b | % Conv. (AH) b | Reactivity Ratio (rEO) c | Mn(GPC) d (kg/mol)/Đ |
|---|---|---|---|---|---|---|
| 1 | SA | 0:1:5:100 | 0 | 0 | - | - |
| 2 | SA | 1:0:5:100 | <1 | 11 | - | - |
| 3 | SA | 1:1.3:5:100 | 88 | 98 | 0.52 | 3.9/1.1 |
| 4 | SA | 1:1.7:5:100 | 100 | 100 | 0.99 | 4.4/1.1 |
| 5 | SA | 1:2.3:5:100 | 100 | 89 | 1.67 | 4.2/1.1 |
| 6 | PA | 1:2.3:5:100 | 89 | 97 | 0.60 | 4.1/1.1 |
| 7 | PA | 1:3.8:5:100 | 100 | 100 | 1.10 | 4.8/1.1 |
| 8 | PA | 1:4.1:5:100 | 100 | 89 | 1.66 | 4.3/1.1 |
| Entry | AH | TBACl:TEB:AH:EO | rEOb | Before Degradation | After Degradation | ||
|---|---|---|---|---|---|---|---|
| Mn (NMR) c (kg/mol) | Mn(GPC) d (kg/mol)/Đ | Mn (NMR) c (kg/mol) | Mn(GPC) d (kg/mol)/Đ | ||||
| 1 | SA | 1:1.3:5:1000 | 0.52 | 44.5 | 36.1/1.2 | 7.22 | 8.21/2.1 |
| 2 | SA | 1:1.7:5:1000 | 0.99 | 44.5 | 38.0/1.2 | 7.60 | 8.34/1.2 |
| 3 | SA | 1:1.7:5:500 | 0.99 | 22.2 | 21.6/1.3 | 4.32 | 4.03/1.3 |
| 4 | SA | 1:2.3:5:1000 | 1.67 | 44.4 | 39.6/1.2 | 8.90 | 9.35/1.9 |
| 5 | PA | 1:2.3:5:1000 | 0.60 | 44.7 | 36.2/1.2 | 7.92 | 8.66/2.2 |
| 6 | PA | 1:3.8:5:1000 | 1.10 | 44.7 | 43.2/1.2 | 8.64 | 8.97/1.3 |
| 7 | PA | 1:4.1:5:1000 | 1.66 | 44.5 | 43.9/1.2 | 9.86 | 10.4/1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chidara, V.K.; Gnanou, Y.; Feng, X. Using Triethylborane to Manipulate Reactivity Ratios in Epoxide-Anhydride Copolymerization: Application to the Synthesis of Polyethers with Degradable Ester Functions. Molecules 2022, 27, 466. https://doi.org/10.3390/molecules27020466
Chidara VK, Gnanou Y, Feng X. Using Triethylborane to Manipulate Reactivity Ratios in Epoxide-Anhydride Copolymerization: Application to the Synthesis of Polyethers with Degradable Ester Functions. Molecules. 2022; 27(2):466. https://doi.org/10.3390/molecules27020466
Chicago/Turabian StyleChidara, Vamshi K., Yves Gnanou, and **aoshuang Feng. 2022. "Using Triethylborane to Manipulate Reactivity Ratios in Epoxide-Anhydride Copolymerization: Application to the Synthesis of Polyethers with Degradable Ester Functions" Molecules 27, no. 2: 466. https://doi.org/10.3390/molecules27020466