
Modulating the Inclusive and Coordinating Ability of Thiacalix[4]arene and Its Antenna Effect on Yb3-Luminescence via Upper-Rim Substitution+
 , , ,
, , , 
Abstract
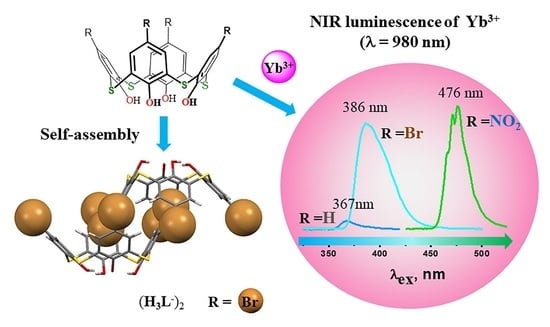
:
1. Introduction
2. Results and Discussion
2.1. UV–Vis Absorption Behavior of H4L(3) and Crystal Structure of H3L(2)−
2.2. Complex Formation of H4L(1–3) with Yb3+ Ions
2.3. Diffusion NMR Spectroscopy
2.4. MALDI-TOF Mass Spectrometry Data
2.5. Computational Modeling of the Yb3+Complexes with p-Nitrothiacalix[4]arene (H4L(3))
2.6. Luminescence Spectroscopy
3. Materials and Methods
3.1. Synthesis of Complex 3 with YbCl3
3.2. Physical Measurements and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Podyachev, S.N.; Zairov, R.R.; Mustafina, A.R. 1,3-Diketone calix [4]arene derivatives—A new type of versatile ligands for metal complexes and nanoparticles. Molecules 2021, 26, 1214. [Google Scholar] [CrossRef] [PubMed]
- Massi, M.; Odgen, M.I.; Ogden, M.I. Luminescent lanthanoid calixarene complexes and materials. Materials 2017, 10, 1369. [Google Scholar] [CrossRef] [Green Version]
- Danil de Namor, A.F.; Cleverley, R.M.; Zapata-Ormachea, M.L. Thermodynamics of Calixarene Chemistry. Chem. Rev. 1998, 98, 2495–2526. [Google Scholar] [CrossRef] [PubMed]
- Padnya, P.; Shibaeva, K.; Arsenyev, M.; Baryshnikova, S.; Terenteva, O.; Shiabiev, I.; Khannanov, A.; Boldyrev, A.; Gerasimov, A.; Grishaev, D.; et al. Catechol-containing schiff bases on thiacalixarene: Synthesis, copper (II) recognition, and formation of organic-inorganic copper-based materials. Molecules 2021, 26, 2334. [Google Scholar] [CrossRef] [PubMed]
- Mandolini, L.; Ungaro, R. >Calixarenes in Action; Mandolini, L., Ungaro, R., Eds.; Imperial College Press: London, UK, 2000; ISBN 978-1-86094-194-8. [Google Scholar]
- Macrocyclic Chemistry: Current Trends and Future Perspectives; Gloe, K. (Ed.) Springer: Dordrecht, The Netherlands, 2005; ISBN 9781402033643. [Google Scholar]
- Podyachev, S.N.; Sudakova, S.N.; Nagimov, R.N.; Lapaev, D.V.; Masliy, A.N.; Syakaev, V.V.; Bazanova, O.B.; Gimazetdinova, G.S.; Babaev, V.M.; Kuznetsov, A.M.; et al. Structural and photophysical properties of Tb3+–Tetra-1,3-diketonate complexes controlled by calix [4]arene-tetrathiacalix [4]arene scaffolds. Dalton Trans. 2019, 48, 3930–3940. [Google Scholar] [CrossRef]
- Podyachev, S.N.; Sudakova, S.N.; Nagimov, R.N.; Masliy, A.N.; Syakaev, V.V.; Lapaev, D.V.; Buzyurova, D.N.; Babaev, V.M.; Gimazetdinova, G.S.; Kuznetsov, A.M.; et al. A simple synthetic approach to enhance the thermal luminescence sensitivity of Tb3+ complexes with thiacalix [4]arene derivatives through upper-rim bromination. Dalton Trans. 2020, 49, 8298–8313. [Google Scholar] [CrossRef] [PubMed]
- Iki, N. Designing strategies for supramolecular luminescent complex of lanthanide–heterometal assembly. Supramol. Chem. 2011, 23, 160–168. [Google Scholar] [CrossRef]
- Tian, H.-W.; Liu, Y.-C.; Guo, D.-S. Assembling features of calixarene-based amphiphiles and supra-amphiphiles. Mater. Chem. Front. 2020, 4, 46–98. [Google Scholar] [CrossRef]
- Desroches, C.; Parola, S.; Vocanson, F.; Perrin, M.; Lamartine, R.; Létoffé, J.-M.; Bouix, J. Nitration of thiacalix [4]arene using nitrosium nitrate complexes: Synthesis and characterization of tetranitro-, tetraamino-, and tetra(4-pyridylimino)tetrahydroxythiacalix [4]arene. New J. Chem. 2002, 26, 651–655. [Google Scholar] [CrossRef]
- Hu, X.; Zhu, Z.; Shen, T.; Shi, X.; Ren, J.; Sun, Q. Synthesis of the tetranitro derivative of thiacalix [4]arene and its acid-base properties. Can. J. Chem. 2004, 82, 1266–1270. [Google Scholar] [CrossRef]
- Hu, X.; Shi, H.; Shi, X.; Zhu, Z.; Sun, Q.; Li, Y.; Yang, H. Selective nitration of thiacalix [4]arene and an investigation of its acid–base properties with a chemometric method. Bull. Chem. Soc. Jpn. 2005, 78, 138–141. [Google Scholar] [CrossRef]
- Muravev, A.; Gerasimova, T.; Fayzullin, R.; Babaeva, O.; Rizvanov, I.; Khamatgalimov, A.; Kadirov, M.; Katsyuba, S.; Litvinov, I.; Latypov, S.; et al. Thermally stable nitrothiacalixarene chromophores: Conformational study and aggregation behavior. Int. J. Mol. Sci. 2020, 21, 6916. [Google Scholar] [CrossRef]
- Liu, Q.; Zou, X.; Shi, Y.; Shen, B.; Cao, C.; Cheng, S.; Feng, W.; Li, F. An efficient dye-sensitized NIR emissive lanthanide nanomaterial and its application in fluorescence-guided peritumoral lymph node dissection. Nanoscale 2018, 10, 12573–12581. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. Lanthanide light for biology and medical diagnosis. J. Lumin. 2016, 170, 866–878. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.-C.G. Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef]
- Nunes, L.R.R.; Labaki, H.P.; Caixeta, F.J.; Gonçalves, R.R. Yb3+ influence on NIR emission from Pr3+-doped spherical yttria nanoparticles for advances in NIR I and NIR II biological windows. J. Lumin. 2022, 241, 118485. [Google Scholar] [CrossRef]
- Kovalenko, A.D.; Pavlov, A.A.; Ustinovich, I.D.; Kalyakina, A.S.; Goloveshkin, A.S.; Marciniak, Ł.; Lepnev, L.S.; Burlov, A.S.; Schepers, U.; Bräse, S.; et al. Highly NIR-emitting ytterbium complexes containing 2-(tosylaminobenzylidene)- N -benzoylhydrazone anions: Structure in solution and use for bioimaging. Dalton Trans. 2021, 50, 3786–3791. [Google Scholar] [CrossRef]
- Ning, Y.; Zhu, M.; Zhang, J.-L. Near-infrared (NIR) lanthanide molecular probes for bioimaging and biosensing. Coord. Chem. Rev. 2019, 399, 213028. [Google Scholar] [CrossRef]
- Kang, X.; Li, C.; Cheng, Z.; Ma, P.; Hou, Z.; Lin, J. Lanthanide—Doped hollow nanomaterials as theranostic agents. WIREs Nanomed. Nanobiotechnol. 2014, 6, 80–101. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Liu, G.; Song, Y.; Li, D.; Dong, X.; Wang, J.; Yu, W. Integrating photoluminescence, magnetism and thermal conversion for potential photothermal therapy and dual-modal bioimaging. J. Colloid Interface Sci. 2018, 510, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Comby, S.; Bünzli, J.-C.G. Lanthanide near-infrared luminescence in molecular probes and devices. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A.J., Bünzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 217–470. [Google Scholar]
- Carnall, W.T. The absorption and fluorescence spectra of rare earth ions in solution. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A.J., Eyring, L., Eds.; Elsevier: Amsterdam, The Netherlands, 1979; pp. 171–208. [Google Scholar]
- Shavaleev, N.M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C.G. Surprisingly bright near-infrared luminescence and short radiative lifetimes of ytterbium in hetero-binuclear Yb−Na chelates. Inorg. Chem. 2009, 48, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Scopelliti, R.; Gumy, F.; Bünzli, J.-C.G. Modulating the near-infrared luminescence of neodymium and ytterbium complexes with tridentate ligands based on benzoxazole-substituted 8-hydroxyquinolines. Inorg. Chem. 2009, 48, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lepnev, L.S.; Utochnikova, V.V. Dual vis-NIR emissive bimetallic naphthoates of Eu–Yb–Gd: A new approach toward Yb luminescence intensity increase through Eu → Yb energy transfer. Phys. Chem. Chem. Phys. 2021, 23, 7213–7219. [Google Scholar] [CrossRef]
- Zairov, R.R.; Dovzhenko, A.P.; Sapunova, A.S.; Voloshina, A.D.; Tatarinov, D.A.; Nizameev, I.R.; Gubaidullin, A.T.; Petrov, K.A.; Enrichi, F.; Vomiero, A.; et al. Dual red-NIR luminescent Eu Yb heterolanthanide nanoparticles as promising basis for cellular imaging and sensing. Mater. Sci. Eng. C 2019, 105, 110057. [Google Scholar] [CrossRef]
- Fedorenko, S.; Gilmanova, D.; Mukhametshina, A.; Nizameev, I.; Kholin, K.; Akhmadeev, B.; Voloshina, A.; Sapunova, A.; Kuznetsova, S.; Daminova, A.; et al. Silica nanoparticles with dual visible–NIR luminescence affected by silica confinement of Tb(III) and Yb(III) complexes for cellular imaging application. J. Mater. Sci. 2019, 54, 9140–9154. [Google Scholar] [CrossRef]
- Iki, N.; Hiro-oka, S.; Nakamura, M.; Tanaka, T.; Hoshino, H. Kinetically stable LnIII complexes comprising a trinuclear core sandwiched between two thiacalix [4]arene ligands self-assembled in water (LnIII = NdIII, YbIII). Eur. J. Inorg. Chem. 2012, 2012, 3541–3545. [Google Scholar] [CrossRef]
- Iki, N.; Tanaka, T.; Hiro-oka, S.; Shinoda, K. Self-assembly of a trilanthanide(III) Ccre sandwiched between two thiacalix [4]arene ligands. Eur. J. Inorg. Chem. 2016, 2016, 5020–5027. [Google Scholar] [CrossRef]
- Karashimada, R.; Iki, N. Thiacalixarene assembled heterotrinuclear lanthanide clusters comprising TbIII and YbIII enable f–f communication to enhance Yb III-centred luminescence. Chem. Commun. 2016, 52, 3139–3142. [Google Scholar] [CrossRef]
- Peng, Y.; Fu, S.; Liu, H.; Lucia, L.A. Accurately determining esterase activity via the isosbestic point of p-nitrophenol. BioResources 2016, 11, 10099–10111. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Y.; Li, H.; Gao, T.; Yan, P. Visible light sensitized near-infrared luminescence of ytterbium via ILCT states in quadruple-stranded helicates. Dalton Trans. 2019, 48, 4026–4034. [Google Scholar] [CrossRef]
- Tsaryuk, V.; Kudryashova, V.; Gawryszewska, P.; Szostak, R.; Vologzhanina, A.; Zhuravlev, K.; Klemenkova, Z.; Legendziewicz, J.; Zolin, V. Structures, luminescence and vibrational spectroscopy of europium and terbium nitro- and dinitro-substituted benzoates. Nitro groups as quenchers of Ln3+ luminescence. J. Photochem. Photobiol. A Chem. 2012, 239, 37–46. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Miao, X.; Deng, W. Halogen bonds fabricate 2D molecular self-assembled nanostructures by scanning tunneling microscopy. Crystals 2020, 10, 1057. [Google Scholar] [CrossRef]
- Puttreddy, R.; Beyeh, N.K.; Ras, R.H.A.; Trant, J.F.; Rissanen, K. Endo-/exo- and halogen-bonded complexes of conformationally rigid C-ethyl-2-bromoresorcinarene and aromatic N-oxides. CrystEngComm. 2017, 19, 4312–4320. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Yan, X.; He, W.; Li, X.; Li, Z.; Mo, Y.; Liu, M.; Jiang, Y.-B. C–I⋯π Halogen bonding driven supramolecular helix of bilateral N-amidothioureas bearing β-Turns. J. Am. Chem. Soc. 2017, 139, 6605–6610. [Google Scholar] [CrossRef]
- Twum, K.; Rissanen, K.; Beyeh, N.K. Recent advances in halogen bonded assemblies with resorcin [4]arenes. Chem. Rec. 2021, 21, 386–395. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Main, M.U.; Pulham, C.R.; Ling, I.; Dalgarno, S.J. A self-assembled nanotube supported by halogen bonding interactions. CrystEngComm 2019, 21, 786–790. [Google Scholar] [CrossRef]
- Ferreira da Rosa, P.P.; Kitagawa, Y.; Hasegawa, Y. Luminescent lanthanide complex with seven-coordination geometry. Coord. Chem. Rev. 2020, 406, 213153. [Google Scholar] [CrossRef]
- Kajiwara, T.; Iki, N.; Yamashita, M. Transition metal and lanthanide cluster complexes constructed with thiacalix[n]arene and its derivatives. Coord. Chem. Rev. 2007, 251, 1734–1746. [Google Scholar] [CrossRef]
- Kajiwara, T.; Katagiri, K.; Hasegawa, M.; Ishii, A.; Ferbinteanu, M.; Takaishi, S.; Ito, T.; Yamashita, M.; Iki, N. Conformation-controlled luminescent properties of lanthanide clusters containing p-tert-butylsulfonylcalix [4]arene. Inorg. Chem. 2006, 45, 4880–4882. [Google Scholar] [CrossRef] [PubMed]
- Altieri, A.S.; Hinton, D.P.; Byrd, R.A. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 1995, 117, 7566–7567. [Google Scholar] [CrossRef]
- Krishnan, V.V. Determination of oligomeric state of proteins in solution from pulsed-field-gradient self-diffusion coefficient measurements. A comparison of experimental, theoretical, and hard-sphere approximated values. J. Magn. Reson. 1997, 124, 468–473. [Google Scholar] [CrossRef]
- Podyachev, S.N.; Sudakova, S.N.; Syakaev, V.V.; Burmakina, N.E.; Shagidullin, R.R.; Morozov, V.I.; Avvakumova, L.V.; Konovalov, A.I. Synthesis and properties of potassium salts of per-O-carboxymethyl-calix [4]pyrogallols and their complexes with Cu2+, Fe3+, and La3+. Russ. Chem. Bull. 2009, 58, 80–88. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Cotton, S.A. Establishing coordination numbers for the lanthanides in simple complexes. Comptes Rendus Chim. 2005, 8, 129–145. [Google Scholar] [CrossRef]
- Shuvaev, S.; Parker, D. A near-IR luminescent ratiometric ytterbium pH probe. Dalton Trans. 2019, 48, 4471–4473. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y. Fluorescent chemo-sensor for metal cations based on thiacalix[4]arenes modified with dansyl moieties at the lower rim. Tetrahedron 2000, 56, 4659–4666. [Google Scholar] [CrossRef]
- Desroches, C.; Lopes, C.; Kessler, V.; Parola, S. Design and synthesis of multifunctional thiacalixarenes and related metal derivatives for the preparation of sol–gel hybrid materials with non-linear optical properties. Dalton Trans. 2003, 2003, 2085–2092. [Google Scholar] [CrossRef]
- Wu, D.; Chen, A.; Johnson, C. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS-2012/1. Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. M.: SHELXTL. Structure Determination Software Suit, SHELXTL, Structure Determination Software Suite, v. 6.1; Bruker AXS Inc.: Madison, WI, USA, 2000. [Google Scholar]
- APEX3. Crystallography Sotware Suite, Version 2018.7-2; BrukerAXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigaku Oxford Diffraction, CrysAlisPro Software system, Version 1.171.42.51 2022; Rigaku Corporation: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A: Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palatinus, L.; Chapuis, G. SUPERFLIP–A computer program for the solution of crystal structures by charge flip** in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Für Krist. Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Diamond—Crystal and Molecular Structure Visualization, Crystal Impact—H. Putz & K. Brandenburg GbR, Kreuzherrenstr. 102, D-53227 Bonn, Germany. Available online: https://www.crystalimpact.de/diamond (accessed on 24 February 2022).
- Schomaker, V.; Trueblood, K.N. On the rigid-body motion of molecules in crystals. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1968, 24, 63–76. [Google Scholar] [CrossRef]
- Lapaev, D.V.; Nikiforov, V.G.; Safiullin, G.M.; Galyaviev, I.G.; Dzabarov, V.I.; Knyazev, A.; Lobkov, V.S.; Galyametdinov, Y. Interligand energy transfer in europium(III) mesogenic adducts. J. Struct. Chem. 2009, 50, 775–781. [Google Scholar] [CrossRef]
- Laikov, D.N. Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem. Phys. Lett. 1997, 281, 151–156. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Laikov, D.N. A new class of atomic basis functions for accurate electronic structure calculations of molecules. Chem. Phys. Lett. 2005, 416, 116–120. [Google Scholar] [CrossRef]
- Bakovets, V.V.; Masliy, A.N.; Kuznetsov, A.M. Formation Thermodynamics of Cucurbit[6]uril Macrocycle Molecules: A Theory Study. J. Phys. Chem. B 2008, 112, 12010–12013. [Google Scholar] [CrossRef] [PubMed]
- Podyachev, S.N.; Gubaidullin, A.T.; Syakaev, V.V.; Sudakova, S.N.; Masliy, A.N.; Saifina, A.F.; Burmakina, N.; Kuznetsov, A.M.; Shagidullin, R.R.; Avvakumova, L.V.; et al. Structural characterization and some coordinational aspects of tetrathiacalix[4]arenes functionalized by hydrazide groups. J. Mol. Struct. 2010, 967, 72–79. [Google Scholar] [CrossRef]
- Podyachev, S.N.; Masliy, A.N.; Semenov, V.E.; Syakaev, V.V.; Sudakova, S.N.; Voronina, J.K.; Ivanov, V.T.; Kuznetsov, A.M.; Gogolashvili, E.L.; Reznik, V.S.; et al. Silver mediated duplex-type complexes of pyrimidinophanes and their acyclic counterparts. RSC Adv. 2015, 5, 16017–16028. [Google Scholar] [CrossRef]
- Kovalenko, E.; Vilaseca, M.; Díaz-Lobo, M.; Masliy, A.N.; Vicent, C.; Fedin, V.P. Supramolecular Adducts of Cucurbit[7]uril and Amino Acids in the Gas Phase. J. Am. Soc. Mass Spectrom. 2015, 27, 265–276. [Google Scholar] [CrossRef]
- Grishaeva, T.N.; Masliy, A.; Kuznetsov, A.M. Water structuring inside the cavities of cucurbit[n]urils (n = 5–8): A quantum-chemical forecast. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 299–313. [Google Scholar] [CrossRef]
- Kovalenko, E.A.; Pashkina, E.A.; Kanazhevskaya, L.Y.; Masliy, A.N.; Kozlov, V.A. Chemical and biological properties of a supramolecular complex of tuftsin and cucurbit[7]uril. Int. Immunopharmacol. 2017, 47, 199–205. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Pollak, P.; Weigend, F. Segmented Contracted Error-Consistent Basis Sets of Double- and Triple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations. J. Chem. Theory Comput. 2017, 13, 3696–3705. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System (Molar Ratio) | Self-Diffusion Coefficients (10−10 m2s−1) | Hydrodynamic Radii rH (Å) |
|---|---|---|
| 2 | 2.32 | 5.5 |
| 2-TEA (1:6) | 2.31 | 5.3 |
| 2- Lu3+-TEA (1:1:6) | 1.95 | 6.6 |
| 3 | 2.37 | 5.4 |
| 3-TEA (1:6) | 2.32 | 5.5 |
| 3- Lu3+-TEA (1:1:6) | 2.04 | 6.3 |
| Reaction | Composition | ΔH0298, kJ | ΔS0298, J/K | ΔG0298, kJ |
|---|---|---|---|---|
| 1 | [YbHL(DMF)4]-(I) | −234.1 | 157.2 | −281.0 |
| 2 | [YbHL(DMF)5]-(II) | −232.8 | 61.3 | −251.1 |
| 3 | [Yb2HL2(DMF)4]-(III) | 41.1 | 217.3 | −23.7 |
| 4 | [Yb2HL2(DMF)4]-(III) | 35.9 | 400.5 | −83.4 |
| 5 | [YbHL(OH)(DMF)3]-(IV) | −193.3 | 157.2 | −240.1 |
| 6 | [Yb2HL2(OH)(DMF)3]−-(V) | −27.0 | 99.7 | −56.7 |
| H4L | Triplet Level, T1 λ, nm (ν, cm−1) at 146 K | <τ> 1 (μs) at 298 K | <τ> 2 (μs) at 146 K |
|---|---|---|---|
| 1 | 417 (23,981) 3 | - | 1437 3 |
| 2 | 458 (21,834) 4 | 24 | 301 4 |
| 3 | 567 (17,637) | 18 | 421 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podyachev, S.N.; Sudakova, S.N.; Zairov, R.R.; Syakaev, V.V.; Masliy, A.N.; Dusek, M.; Gubaidullin, A.T.; Dovzhenko, A.P.; Buzyurova, D.N.; Lapaev, D.V.;
et al. Modulating the Inclusive and Coordinating Ability of Thiacalix[4]arene and Its Antenna Effect on Yb3-Luminescence via Upper-Rim Substitution
Podyachev SN, Sudakova SN, Zairov RR, Syakaev VV, Masliy AN, Dusek M, Gubaidullin AT, Dovzhenko AP, Buzyurova DN, Lapaev DV,
et al. Modulating the Inclusive and Coordinating Ability of Thiacalix[4]arene and Its Antenna Effect on Yb3-Luminescence via Upper-Rim Substitution
Podyachev, Sergey N., Svetlana N. Sudakova, Rustem R. Zairov, Victor V. Syakaev, Alexey N. Masliy, Michal Dusek, Aidar T. Gubaidullin, Alexey P. Dovzhenko, Daina N. Buzyurova, Dmitry V. Lapaev,
and et al. 2022. "Modulating the Inclusive and Coordinating Ability of Thiacalix[4]arene and Its Antenna Effect on Yb3-Luminescence via Upper-Rim Substitution