
Coinage Metal-Catalyzed Asymmetric Reactions of ortho-Alkynylaryl and Heteroaryl Aldehydes and Ketones

Abstract
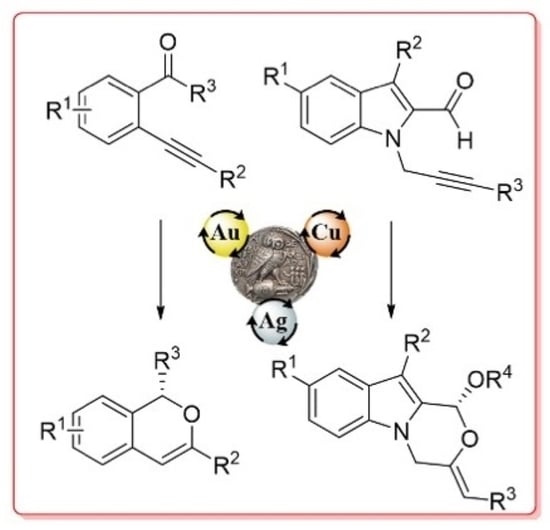
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gold Catalysis
3. Silver Catalysis
4. Copper Catalysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aubert, C.; Buisine, O.; Malacria, M. The Behavior of 1,n-Enynes in the Presence of Transition Metals. Chem. Rev. 2002, 102, 813–834. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, G.C. Mechanistic Aspects of Transition Metal Catalysed 1,6-Diene and 1,6-Enyne Cycloisomerisation Reactions. Org. Biomol. Chem. 2003, 1, 215–236. [Google Scholar] [CrossRef]
- Stephen, A.; Hashmi, K. Homogeneous Catalysis by Gold. Gold Bull. 2004, 37, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sun, J.; Kozmin, S.A. Gold and Platinum Catalysis of Enyne Cycloisomerization. Adv. Synth. Catal. 2006, 348, 2271–2296. [Google Scholar] [CrossRef]
- Michelet, V.; Toullec, P.Y.; Genêt, J.-P. Cycloisomerization of 1,n-Enynes: Challenging Metal-Catalyzed Rearrangements and Mechanistic Insights. Angew. Chem. Int. Ed. 2008, 47, 4268–4315. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Núñez, E.; Echavarren, A.M. Gold-Catalyzed Cycloisomerizations of Enynes: A Mechanistic Perspective. Chem. Rev. 2008, 108, 3326–3350. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C. Recent Advances in Syntheses of Carbocycles and Heterocycles via Homogeneous Gold Catalysis. Part 2: Cyclizations and Cycloadditions. Tetrahedron 2008, 64, 7847–7870. [Google Scholar] [CrossRef]
- Belmont, P.; Parker, E. Silver and Gold Catalysis for Cycloisomerization Reactions. Eur. J. Org. Chem. 2009, 2009, 6075–6089. [Google Scholar] [CrossRef]
- Lee, S.I.; Chatani, N. Catalytic Skeletal Reorganization of Enynes through Electrophilicactivation of Alkynes: Double Cleavage of C–C Double and Triple Bonds. Chem. Commun. 2009, 45, 371–384. [Google Scholar] [CrossRef]
- Toullec, P.Y.; Michelet, V. Cycloisomerization of 1,n-Enynes Via Carbophilic Activation. Top Curr. Chem. 2011, 302, 31–80. [Google Scholar] [CrossRef]
- Krause, N.; Winter, C. Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carbo-and Heterocycles. Chem. Rev. 2011, 111, 1994–2009. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Zhang, Z.; Shi, M. Transition Metal-Catalyzed Carbocyclization of Nitrogen and Oxygen-Tethered 1,n-Enynes and Diynes: Synthesis of Five or Six-Membered Heterocyclic Compounds. Chem. Commun. 2012, 48, 10271. [Google Scholar] [CrossRef] [PubMed]
- Kazem Shiroodi, R.; Gevorgyan, V. Metal-Catalyzed Double Migratory Cascade Reactions of Propargylic Esters and Phosphates. Chem. Soc. Rev. 2013, 42, 4991. [Google Scholar] [CrossRef] [PubMed]
- Pflästerer, D.; Hashmi, A.S.K. Gold Catalysis in Total Synthesis–Recent Achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef] [PubMed]
- Joost, M.; Amgoune, A.; Bourissou, D. Reactivity of Gold Complexes towards Elementary Organometallic Reactions. Angew. Chem. Int. Ed. 2015, 54, 15022–15045. [Google Scholar] [CrossRef] [PubMed]
- Asao, N.; Nogami, T.; Takahashi, K.; Yamamoto, Y. Pd(II) Acts Simultaneously as a Lewis Acid and as a Transition-Metal Catalyst: Synthesis of Cyclic Alkenyl Ethers from Acetylenic Aldehydes. J. Am. Chem. Soc. 2002, 124, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Nogami, T.; Asao, N.; Yamamoto, Y. Synthesis of Novel Antitumor Agent 1-methoxy-5,10-dioxo-5,10-dihydro-1H-benzo[g]isochromene Carboxylic Acid (3-Dimethylylaminopropyl)amide with a Dual Role Pd(II) Catalyst. J. Org. Chem. 2003, 68, 9496–9498. [Google Scholar] [CrossRef]
- Yamamoto, Y. From σ- to π-Electrophilic Lewis Acids. Application to Selective Organic Transformations. J. Org. Chem. 2007, 72, 7817–7831. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Singh, B.; Khanna, R.S.; Singh, R.M. Copper-Free Palladium-Catalyzed Sonogashira Coupling−Annulation: Efficient One-Pot Synthesis of Functionalized Pyrano [4,3-b]Quinolines from 2-Chloro-3-Formylquinolines. J. Org. Chem. 2009, 74, 5664–5666. [Google Scholar] [CrossRef]
- Nardangeli, N.; Topolovčan, N.; Simionescu, R.; Hudlický, T. Polarization Effect on Regioselectivity of Pd-Catalyzed Cyclization of 2-Alkynylbenzaldehydes. Eur. J. Org. Chem. 2020, 2020, 227–233. [Google Scholar] [CrossRef]
- Singh, R.M.; Kumar, R.; Bharadwaj, K.C.; Gupta, T. Pd Catalyzed Facile Synthesis of Cyclopenta[b]Quinolin-1-One via Sequential Sonogashira Coupling and Annulation. An Unusual Mode of Ring Closure, Using Sulphur as a Soft Nucleophile. Org. Chem. Front. 2016, 3, 1100–1104. [Google Scholar] [CrossRef]
- Bacchi, A.; Costa, M.; Della Cà, N.; Fabbricatore, M.; Fazio, A.; Gabriele, B.; Nasi, C.; Salerno, G. Synthesis of 1-(Alkoxycarbonyl)Methylene-1,3-Dihydroisobenzofurans and 4-(Alkoxycarbonyl)Benzo[c]Pyrans by Palladium-Catalysed Oxidative Carbonylation of 2-Alkynylbenzyl Alcohols, 2-Alkynylbenzaldehydes and 2-Alkynylphenyl Ketones. Eur. J. Org. Chem. 2004, 574–585. [Google Scholar] [CrossRef]
- Saxena, A.; Perez, F.; Krische, M.J. Ruthenium(0)-Catalyzed [4+2] Cycloaddition of Acetylenic Aldehydes with α-Ketols: Convergent Construction of Angucycline Ring Systems. Angew. Chem. 2016, 128, 1515–1519. [Google Scholar] [CrossRef]
- Ruch, A.A.; Kong, F.; Nesterov, V.N.; Slaughter, L.M. Tetracyclic Dihydronaphthalene Derivatives via Gold-Catalyzed Aminative Homodimerization of ortho-Alkynylbenzaldehydes. Chem. Commun. 2016, 52, 14133–14136. [Google Scholar] [CrossRef] [PubMed]
- Kotera, A.; Uenishi, J.; Uemura, M. Gold-Catalyzed Stereoselective Reaction of Tricarbonylchromium Complexes of ortho-Alkynyl Benzaldehydes and Benzaldimines with Nucleophiles. J. Organomet. Chem. 2010, 695, 2180–2190. [Google Scholar] [CrossRef]
- Kotera, A.; Uenishi, J.; Uemura, M. Gold(I)-Catalyzed Stereoselective Cyclization of Ortho Alkynyl Benzaldehyde Chromium Complexes with Nucleophiles. Tetrahedron Lett. 2010, 51, 1166–1169. [Google Scholar] [CrossRef]
- Dell’Acqua, M.; Facoetti, D.; Abbiati, G.; Rossi, E. Selective Base-Promoted Synthesis of Dihydroisobenzofurans by Domino Addition/Annulation Reactions of ortho-Alkynylbenzaldehydes. Synthesis 2010, 2010, 2367–2378. [Google Scholar] [CrossRef]
- Garanzini, D.; Pirovano, V.; Menghi, I.; Celentano, G.; Rizzato, S.; Rossi, E.; Caselli, A.; Abbiati, G. [Ag(PcL)]-Catalysed Domino Approach to 6-Substituted Benzoxazino Isoquinolines. Eur. J. Org. Chem. 2020, 2020, 3660–3670. [Google Scholar] [CrossRef]
- Pirovano, V.; Hamdan, G.; Garanzini, D.; Brambilla, E.; Rossi, E.; Caselli, A.; Abbiati, G. [Ag(PcL)]-Catalyzed Domino Reactions of 2-Alkynylbenzaldehydes with Electron-Poor Anilines: Synthesis of 1-Aminoisochromenes. Eur. J. Org. Chem. 2020, 2020, 2592–2599. [Google Scholar] [CrossRef]
- Rustagi, V.; Tiwari, R.; Verma, A.K. AgI-Catalyzed Cascade Strategy: Regioselective Access to Diversely Substituted Fused Benzimidazo [2,1-a]Isoquinolines, Naphthyridines, Thienopyridines, and Quinoxalines in Water. Eur. J. Org. Chem. 2012, 2012, 4590–4602. [Google Scholar] [CrossRef]
- Yuan, B.; He, R.; Shen, W.; Huang, C.; Li, M. Mechanistic Insights into the Cu(I)- and Cu(II)-Catalyzed Cyclization of o-Alkynylbenzaldehydes: The Solvent DMF and Oxidation State of Copper Affect the Reaction Mechanism. J. Org. Chem. 2015, 80, 6553–6563. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Yamamoto, Y. Synthesis of Cyclic Alkenyl Ethers via Intramolecular Cyclization of O-Alkynylbenzaldehydes. Importance of Combination between CuI Catalyst and DMF. J. Org. Chem. 2004, 69, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 18, 734–736. [Google Scholar] [CrossRef]
- Zi, W.; Dean Toste, F. Recent Advances in Enantioselective Gold Catalysis. Chem. Soc. Rev. 2016, 45, 4567–4589. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Enantioselective Silver-Catalyzed Transformations. Chem. Rev. 2016, 116, 14868–14917. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Das, B.G.; Singh, V.K. Recent Advancement in Copper-Catalyzed Asymmetric Reactions of Alkynes. Tetrahedron 2021, 93, 132238. [Google Scholar] [CrossRef]
- Yao, X.; Li, C.-J. Water-Triggered and Gold(I)-Catalyzed Cascade Addition/Cyclization of Terminal Alkynes with ortho-Alkynylaryl Aldehyde. Org. Lett. 2006, 8, 1953–1955. [Google Scholar] [CrossRef]
- Obika, S.; Kono, H.; Yasui, Y.; Yanada, R.; Takemoto, Y. Concise Synthesis of 1,2-Dihydroisoquinolines and 1H-Isochromenes by Carbophilic Lewis Acid-Catalyzed Tandem Nucleophilic Addition and Cyclization of 2-(1-Alkynyl)Arylaldimines and 2-(1-Alkynyl)Arylaldehydes. J. Org. Chem. 2007, 72, 4462–4468. [Google Scholar] [CrossRef]
- Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Silver versus Gold Catalysis in Tandem Reactions of Carbonyl Functions onto Alkynes: A Versatile Access to Furoquinoline and Pyranoquinoline Cores. Chem. Eur. J. 2007, 13, 5632–5641. [Google Scholar] [CrossRef]
- Liu, L.-P.; Hammond, G.B. Gold-Catalyzed Cascade Annulations of 2-(Ynol)Aryl Aldehydes: Facile Synthesis of Benzochromanes and Benzobicyclo[n.3.1]Acetals. Org. Lett. 2010, 12, 4640–4643. [Google Scholar] [CrossRef]
- Tomás-Mendivil, E.; Heinrich, C.F.; Ortuno, J.-C.; Starck, J.; Michelet, V. Gold-Catalyzed Access to 1H-Isochromenes: Reaction Development and Mechanistic Insight. ACS Catal. 2017, 7, 380–387. [Google Scholar] [CrossRef]
- Michalska, M.; Grudzień, K.; Małecki, P.; Grela, K. Gold(I)-Catalyzed Formation of Naphthalene/Acenaphthene Heterocyclic Acetals. Org. Lett. 2018, 20, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Handa, S.; Slaughter, L.M. Enantioselective Alkynylbenzaldehyde Cyclizations Catalyzed by Chiral Gold(I) Acyclic Diaminocarbene Complexes Containing Weak Au-Arene Interactions. Angew. Chem. Int. Ed. 2012, 51, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.-F.; Ko, H.-M.; Shing, K.-P.; Deng, J.-R.; Lai, N.C.-H.; Wong, M.-K. C,O-Chelated BINOL/Gold(III) Complexes: Synthesis and Catalysis with Tunable Product Profiles. Angew. Chem. Int. Ed. 2017, 56, 3074–3079. [Google Scholar] [CrossRef] [PubMed]
- Zi, W.; Toste, F.D. Gold(I)-Catalyzed Enantioselective Carboalkoxylation of Alkynes. J. Am. Chem. Soc. 2013, 135, 12600–12603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.-J.; Cui, J.-F.; Yang, B.; Ning, Y.; Lai, N.C.-H.; Wong, M.-K. Chiral Cyclometalated Oxazoline Gold(III) Complex-Catalyzed Asymmetric Carboalkoxylation of Alkynes. Org. Lett. 2019, 21, 6289–6294. [Google Scholar] [CrossRef] [PubMed]
- Dupeux, A.; Michelet, V. Gold-Catalyzed Domino Cycloisomerization/Alkoxylation: An Entry to 3,4-Dihydro-1H-[1,4]Oxazino [4,3-a]Indole. J. Org. Chem. 2021, 86, 17738–17747. [Google Scholar] [CrossRef]
- Terada, M.; Li, F.; Toda, Y. Chiral Silver Phosphate Catalyzed Transformation of Ortho -Alkynylaryl Ketones into 1H-Isochromene Derivatives through an Intramolecular-Cyclization/Enantioselective-Reduction Sequence. Angew. Chem. Int. Ed. 2014, 53, 235–239. [Google Scholar] [CrossRef]
- Bröhl, N.; Kundu, D.; Raabe, G.; Enders, D. One-Pot Synthesis of 1-Substituted 1H-Isochromenes by Combining Brønsted Acid with Silver Catalysis. Synthesis 2016, 49, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Chen, Y.; Cao, H.; Wei, Q.; Yang, Y.; Ouyang, Q.; Peng, Y. Asymmetric Cyclization/Nucleophilic Tandem Reaction of o-Alkynylacetophenone with (Diazomethyl)Phosphonate for the Synthesis of Functional Isochromenes. Org. Lett. 2019, 21, 7597–7601. [Google Scholar] [CrossRef]
- Saito, K.; Kajiwara, Y.; Akiyama, T. Chiral Copper(II) Phosphate Catalyzed Enantioselective Synthesis of Isochromene Derivatives by Sequential Intramolecular Cyclization and Asymmetric Transfer Hydrogenation of o-Alkynylacetophenones. Angew. Chem. Int. Ed. 2013, 52, 13284–13288. [Google Scholar] [CrossRef] [PubMed]
- Miao, T.; Tian, Z.-Y.; He, Y.-M.; Chen, F.; Chen, Y.; Yu, Z.-X.; Fan, Q.-H. Asymmetric Hydrogenation of In Situ Generated Isochromenylium Intermediates by Copper/Ruthenium Tandem Catalysis. Angew. Chem. Int. Ed. 2017, 56, 4135–4139. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.P.; Bodiwala, H.S. Recent Advances in Anti-HIV Natural Products. Nat. Prod. Rep. 2010, 27, 1781. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-M.; Yang, S.-X.; Qin, J.-C. Azaphilones: Chemistry and Biology. Chem. Rev. 2013, 113, 4755–4811. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.E.; Chih-Chien Cheng, K.; Wei, W.-G.; Yuan, P.; Dai, P.; Trilles, R.; Ni, F.; Yuan, J.; MacArthur, R.; Guha, R.; et al. Discovery of New Antimalarial Chemotypes through Chemical Methodology and Library Development. Proc. Natl. Acad. Sci. USA 2011, 108, 6775–6780. [Google Scholar] [CrossRef] [Green Version]
- Trisuwan, K.; Khamthong, N.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Anthraquinone, Cyclopentanone, and Naphthoquinone Derivatives from the Sea Fan-Derived Fungi Fusarium spp. PSU-F14 and PSU-F135. J. Nat. Prod. 2010, 73, 1507–1511. [Google Scholar] [CrossRef]
- Shishido, Y.; Wakabayashi, H.; Koike, H.; Ueno, N.; Nukui, S.; Yamagishi, T.; Murata, Y.; Naganeo, F.; Mizutani, M.; Shimada, K.; et al. Discovery and Stereoselective Synthesis of the Novel Isochroman Neurokinin-1 Receptor Antagonist ‘CJ-17,493. ’ Bioorg. Med. Chem. 2008, 16, 7193–7205. [Google Scholar] [CrossRef]
- Liu, M.; **ao, H.-T.; He, H.; Hao, X.-Y. A Novel Lignanoid and Norbisabolane Sesquiterpenoids from Glochidion Puberum. Chem. Nat. Compd. 2008, 44, 588–590. [Google Scholar] [CrossRef]
- Tamanna; Kumar, M.; Joshi, K.; Chauhana, P. Catalytic Asymmetric Synthesis of Isochroman Derivatives. Adv. Synth. Catal. 2020, 362, 1907–1926. [Google Scholar] [CrossRef]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melot, R.; Michelet, V. Coinage Metal-Catalyzed Asymmetric Reactions of ortho-Alkynylaryl and Heteroaryl Aldehydes and Ketones. Molecules 2022, 27, 6970. https://doi.org/10.3390/molecules27206970
Melot R, Michelet V. Coinage Metal-Catalyzed Asymmetric Reactions of ortho-Alkynylaryl and Heteroaryl Aldehydes and Ketones. Molecules. 2022; 27(20):6970. https://doi.org/10.3390/molecules27206970
Chicago/Turabian StyleMelot, Romain, and Véronique Michelet. 2022. "Coinage Metal-Catalyzed Asymmetric Reactions of ortho-Alkynylaryl and Heteroaryl Aldehydes and Ketones" Molecules 27, no. 20: 6970. https://doi.org/10.3390/molecules27206970