1. Introduction
Organic dye molecules find a wide range of applications in optoelectronics and photonics, from active media in lasers [
1], to fluorescent labels in confocal microscopy [
2] and light emitters in displays [
3]. Besides these technological applications, dye molecules are also of great interest for fundamental research studies. In recent years, single organic molecules have witnessed an increasing use in quantum technologies due to their favorable properties such as: small size, easy spectral tunability thanks to chemical design and synthesis, high photoluminescence quantum efficiency and high degree of coherence when cooled to low temperatures [
4]. When placed inside an optical microcavity, upon strong coupling between excitonic and photonic modes, organic dyes have also been shown to form cavity polaritons [
5,
6], which are hybrid light—matter quasi particles. The lifetime of such hybrid states is defined both by the rate at which photons ‘leak’ from the cavity, and by the rate at which light and matter mutually dephase. Thus, the choice of molecular materials having long dephasing times may allow for enhanced polariton lifetimes, thereby facilitating the build-up of polariton populations and the emergence of non-linear phenomena. For example, the recently demonstrated polariton condensation [
7] and super-extensive light absorption important for the development of Dicke quantum battery [
8]. In summary, both cavity polaritonics and the development of quantum technologies, in which information is imprinted on a single molecule in the form of an excitonic qubit, critically require the characterization of the dephasing processes in dye molecules.
Organic dye molecules can be described with different models based on their complexity [
9,
10]; in this work, the dye under study can be approximated to the first order of as a two-level system (TLS), with the energy of the transition from the ground (
) state to the excited (
) state,
, falling in the visible range. Upon excitation of a TLS, a quantum coherent superposition of ground and excited states is created, described by the wave function
, with
and
the probability of finding the system in the ground and excited state, respectively. Interactions with the environment cause the decay of this superposition (decoherence processes), projecting the wave function on either the ground or the excited state. For a collection of TLSs, such as dye molecules in solution or in a film, the quantum state is more appropriately represented by the density operator
, with its off-diagonal elements
describing collective coherences and the diagonal terms
and
describing populations of the states [
11]. In this case, dynamic interactions with the environment cause the decay of the macroscopic polarization, which can be phenomenologically described within the Bloch equation [
11]. For an ensemble of TLSs all with the same transition energy
, the absorption spectral profile is Lorentzian with linewidth inversely proportional to the dephasing time
, and the system is said to be homogeneously broadened. Moreover, the TLS can have a distribution of transition energies
, often described by a Gaussian function, which, combined with dynamic perturbations, describes a situation known as inhomogeneous broadening. In this case, the absorption spectrum is the convolution between the Lorentzian linewidths of the individual transitions and the Gaussian distribution of their energies, resulting in the so-called Voigt profile.
Several nonlinear spectroscopy techniques have been used to measure the dephasing time of molecular ensembles. In the frequency domain, spectral hole burning [
12] consists of the use of an intense narrowband pump pulse to excite the molecules with transition frequencies close to the pump pulse frequency, thus saturating these transitions and burning a hole in the absorption spectrum. This is then detected by a broadband probe pulse), with spectral width corresponding to the homogeneous linewidth. The use of a narrowband pump pulse, however, unavoidably results in the loss of temporal resolution. In the time domain, non-collinear degenerate four-wave-mixing spectroscopies such as two- and three-pulse photon echo (2PPE/3PPE) [
13] enable dephasing times to be determined with the highest possible temporal resolution. In photon echo spectroscopies, a first pulse creates a polarization in the sample, and a second non-collinear pulse, delayed by the coherence time t
1, interferes with this polarization, generating a population grating which diffracts either the second pulse itself (in 2PPE) or a third pulse delayed by the so-called population or waiting time t
2 (in 3PPE), resulting in the emission of a third-order nonlinear signal E
(3)(t
1, t
2, t
3), where t
3 is the detection time. In 2PPE/3PPE the diffracted signal is integrated in time, and its energy is measured as a function of the delay t
1, with the signal decay time constant providing a direct measurement of the dephasing time. Often, when the decay time of the signal is comparable to or shorter than the duration of the excitation pulses, one uses the 3PPE peak shift approach [
14], which consists of measuring the temporal delay between the maxima of the 3PPE signals emitted in different phase-matched directions.
Two-dimensional electronic spectroscopy (2DES) [
15,
16] is an advanced version of 3PPE in which the third-order nonlinear signal emitted by the sample following excitation by the three pulses is fully measured in amplitude and phase by spectral interferometry with a fourth pulse, the local oscillator (LO), which is phase-locked to the third pulse. The nonlinear signal is usually spectrally resolved by a spectrometer that performs a Fourier transform (FT) with respect to the detection time t
3, to obtain the detection energy axis
. By performing an additional FT of the signal with respect to the coherence time t
1, which requires interferometric stability of this delay as well, one obtains the excitation energy axis
, with the final dataset corresponding to a 2D map correlating excitation (
) and detection (
) energies for a fixed value of the waiting time t
2. Thanks to the FT approach, 2DES simultaneously combines very high temporal and spectral resolution. The diagonal peaks in the purely absorptive 2DES maps provide the most direct information on homogeneous and inhomogeneous broadening and how they evolve in time, i.e., on spectral diffusion processes [
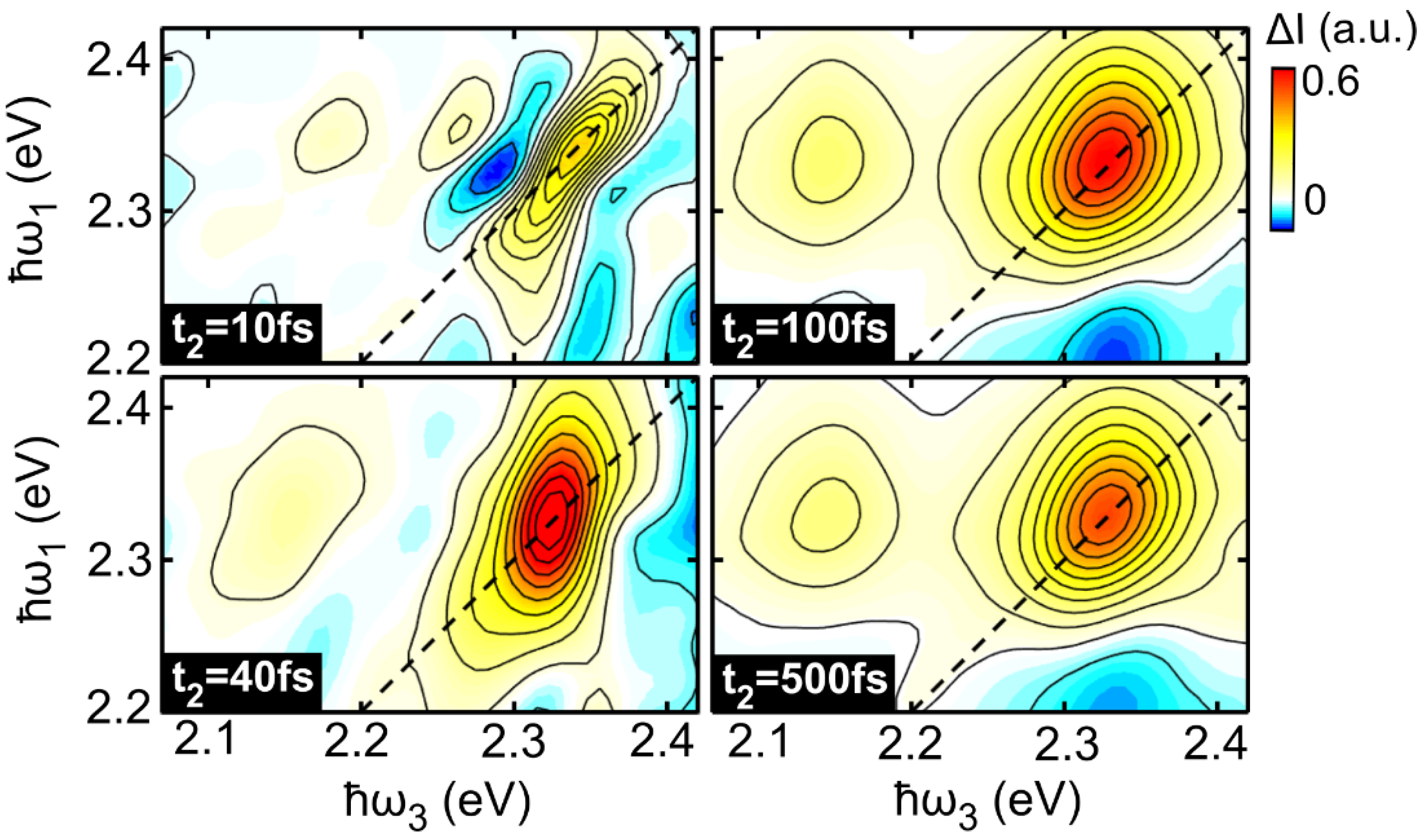
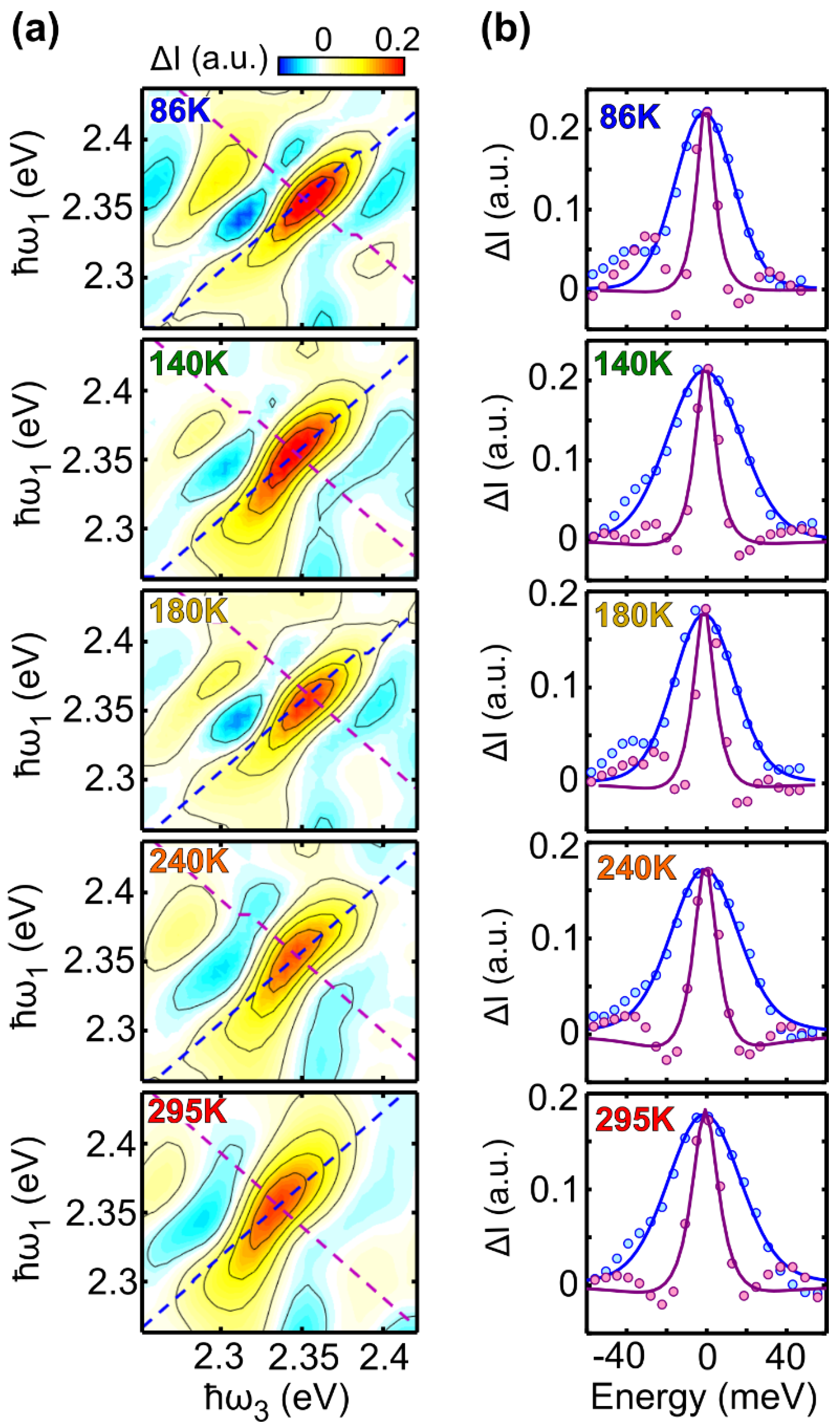
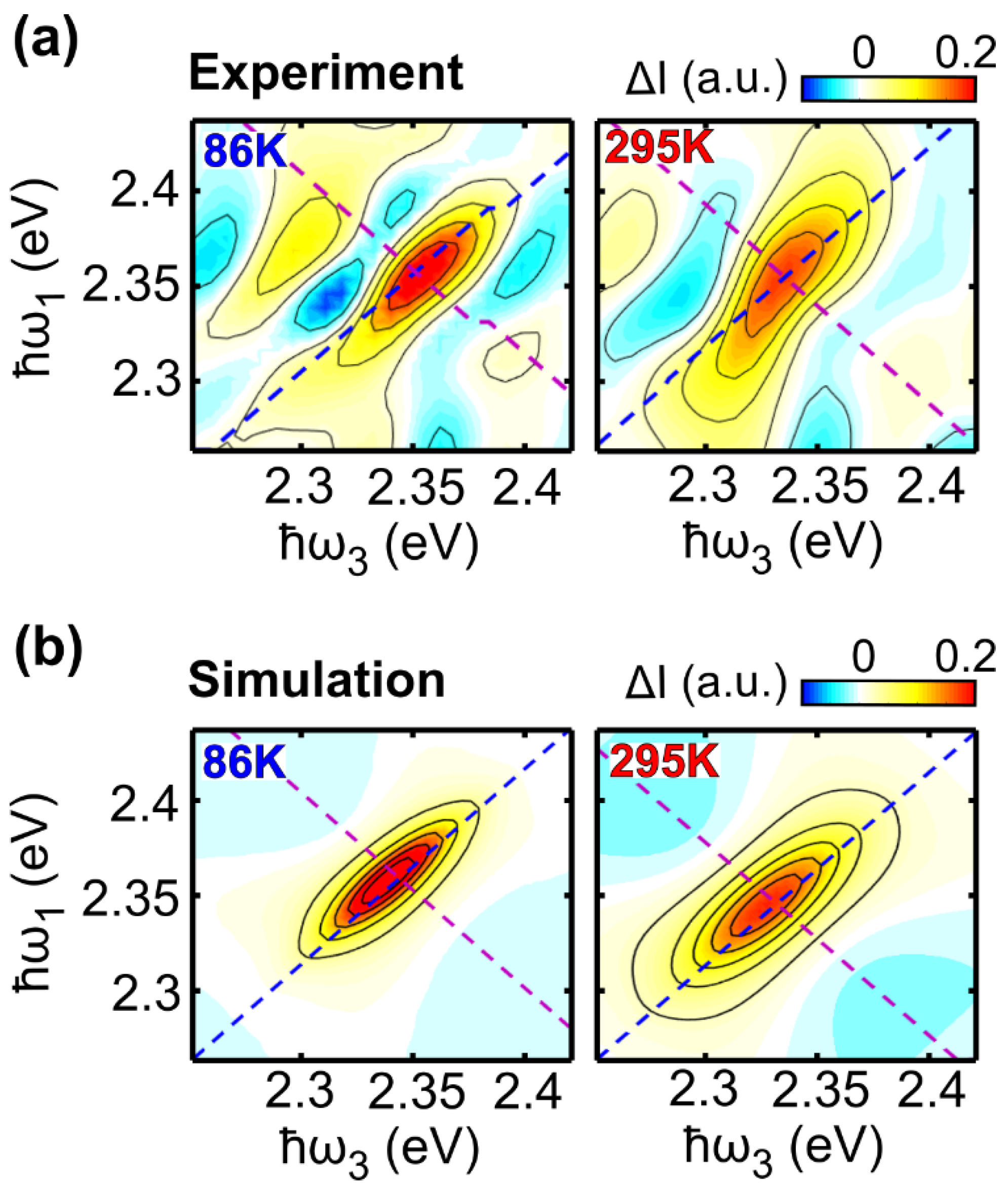
17]. In the case of a collection of TLSs with different transition energies (large inhomogeneous broadening limit) the 2DES peak will appear strongly elongated along the excitation axis, while for pure homogeneous broadening the peak will be symmetric. When the correlation between excitation and detection frequencies is lost, as in the case of spectral diffusion, the peak becomes progressively more symmetric.
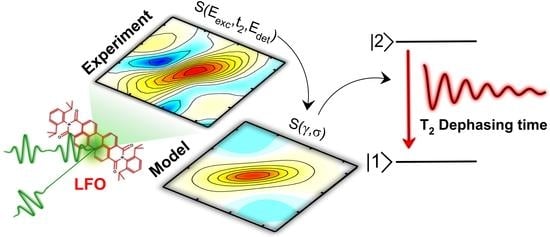
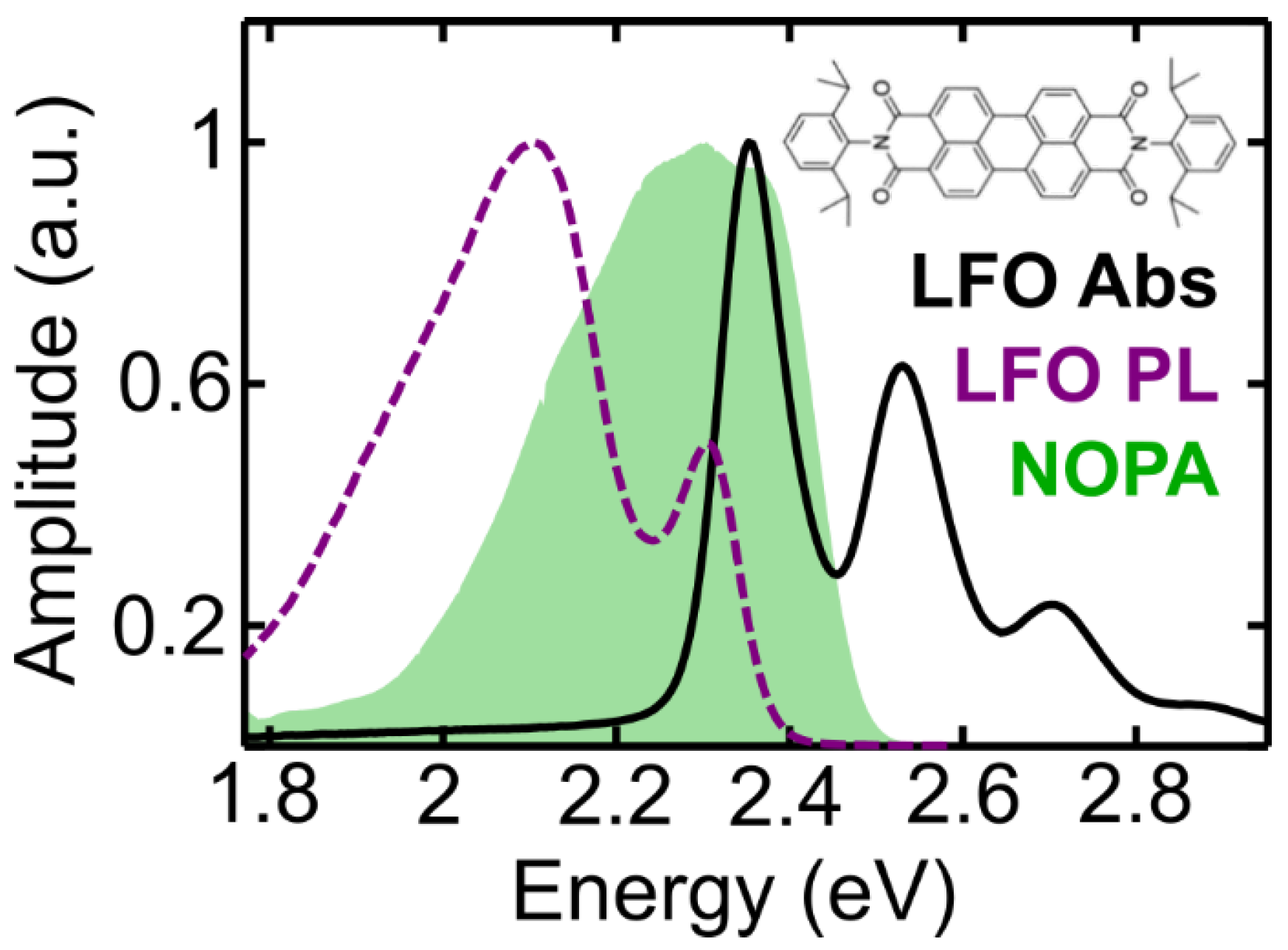
In this paper we exploit 2DES to study the dephasing processes in the prototypical small dye molecule Lumogen-F orange (LFO), which was recently shown to undergo super-absorption when embedded in a microcavity [
8]. We study LFO in solution and embedded in a polymer matrix, and in the latter case we perform a temperature dependent study. We introduce a model that simultaneously fits the homogeneous and the inhomogeneous broadening and extracts the dephasing time from the absorptive 2DES maps. Finally, we use our model to describe the spectral diffusion dynamics at different temperatures and to reconstruct the steady-state absorption spectrum of the molecule.
4. Conclusions
Characterizing decoherence mechanisms in molecular dyes is important for their application in photonics and quantum technologies. Conventional spectroscopic techniques use multiple approaches to determine dephasing times and spectral diffusion processes [
12,
14,
19,
20,
41,
42]. Conversely, 2DES is an ideal tool for retrieving such information all together; it simultaneously differentiates the contributions from homogeneous and inhomogeneous broadening mechanisms by the shape of the diagonal peaks, while following the loss of excitation memory due to spectral diffusion, thanks to its combination of high temporal and spectral resolution.
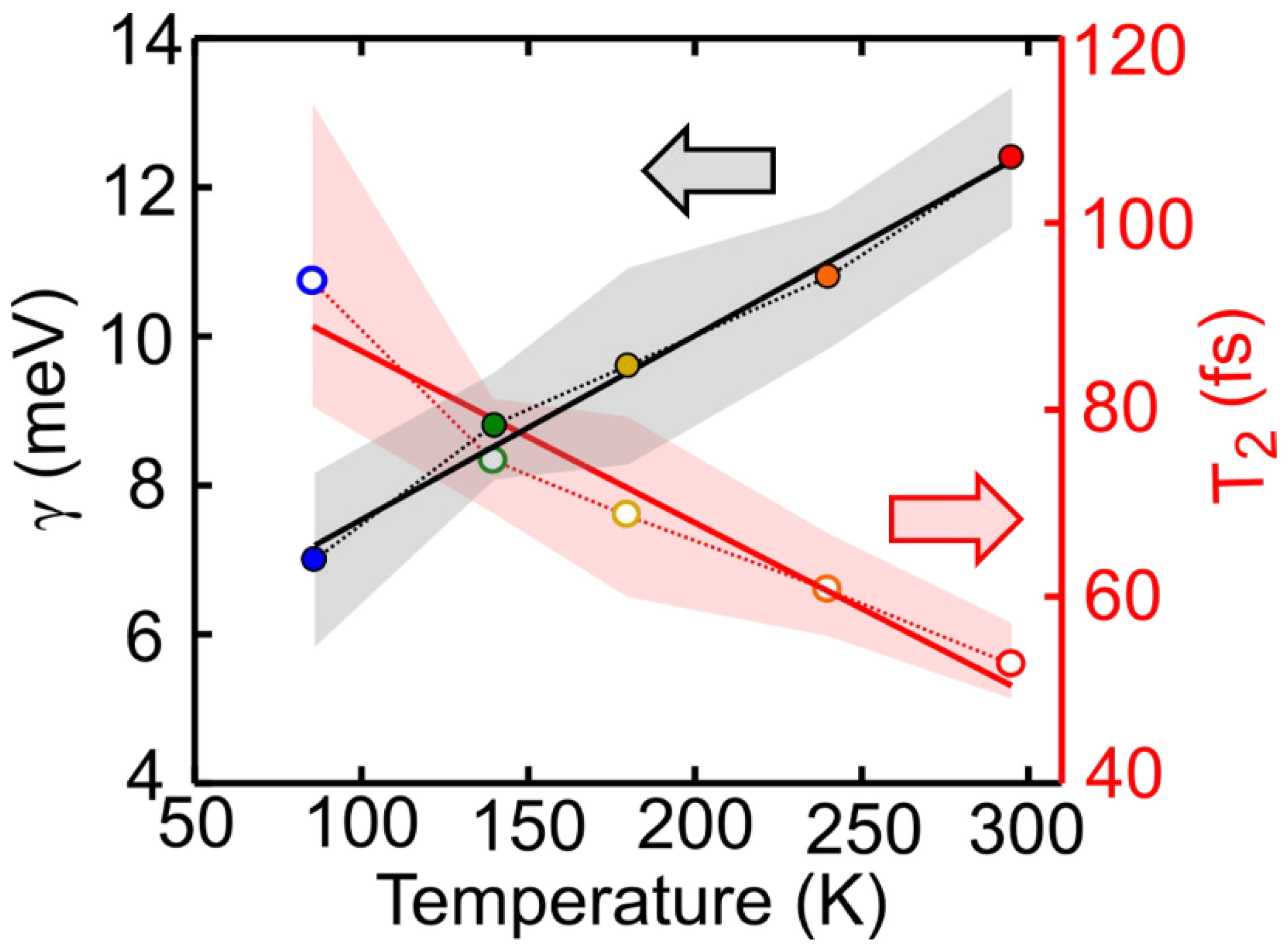
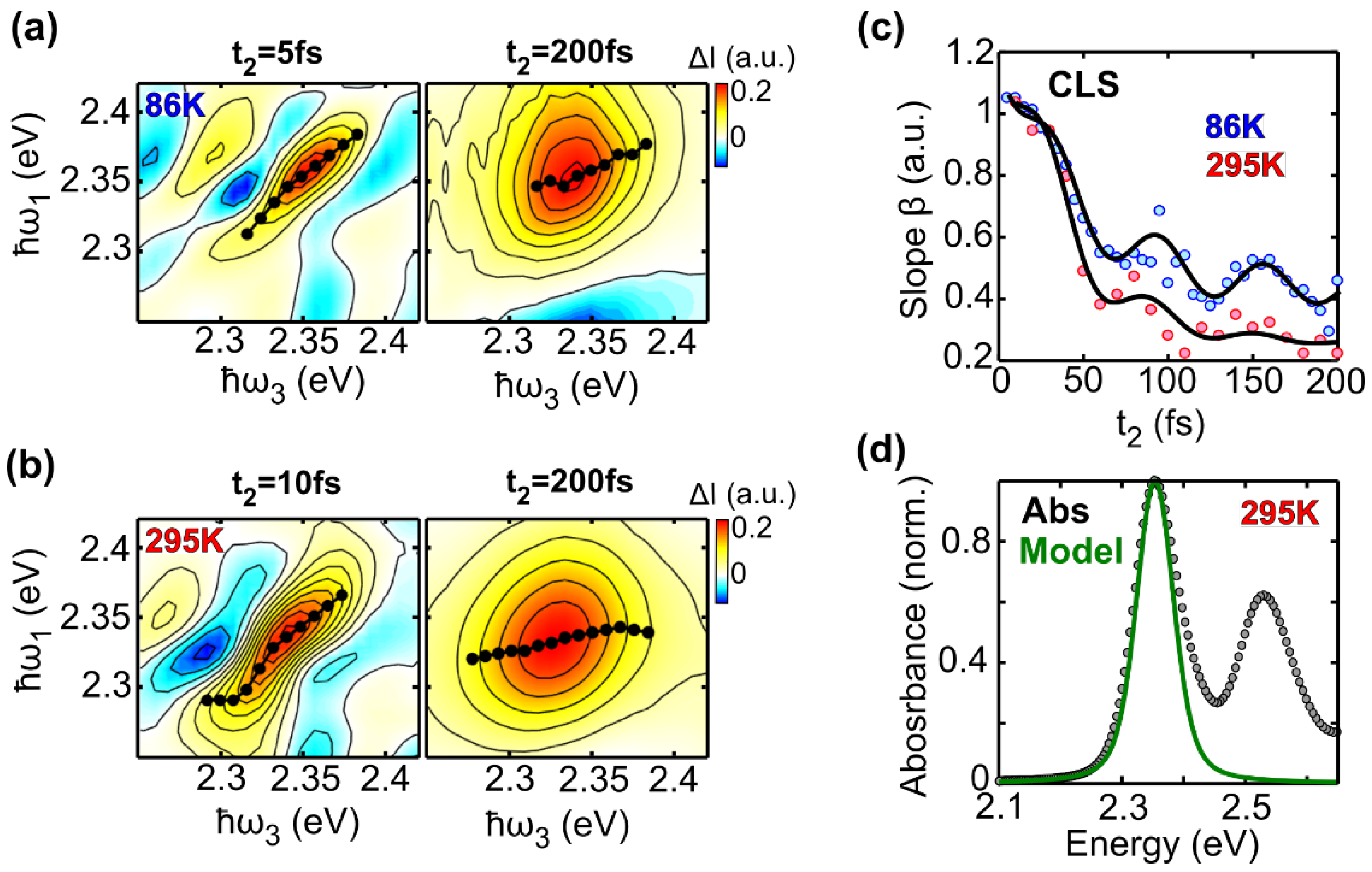
In this study we have performed 2DES of the prototypical molecular dye LFO embedded in a polymer under low aggregation conditions, which allows intermolecular interactions to be neglected. We accurately reproduced the experimental 2DES maps via a model using the Bloch equations for a two-level system. At 295 K we obtained a dephasing rate of γ = 12.4 meV, which corresponds to a dephasing time of T2 = 53 fs. A similar result is obtained for the molecule diluted in DCM (γ = 11.5 meV), and by applying an inhomogeneous limit model (γ = 12.7 meV). Temperature dependent 2DES measurements from 86 K to 295 K show a linear dependence of the dephasing rate on the temperature; at T = 86 K, the dephasing rate decreases to γ = 7 meV, with a dephasing time T2 = 94 fs, which is nearly doubled with respect to room temperature. Measurements as a function of the waiting time show a rapid loss of excitation memory on the 200 fs timescale due to spectral diffusion, with the CLS of the peaks modeled with underdamped and overdamped Brownian oscillators combined with an ultrafast decay.
Overall, the results of our 2DES experiments in LFO are in good agreement with previous studies of dephasing processes in organic dyes. We believe that our results will provide useful parameters for the design of advanced optoelectronic and photonic devices (such as polaritonic cavities and organic quantum batteries) exploiting such molecules.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}