1. Introduction
Herpes simplex virus type 2 (HSV-2) is considered to be one of the most severe pathogens in humans causing an important sexually transmitted disease known as genital herpes. Notably, infection with HSV-2 has been declared high risk for HIV infection, as well as invasive cervical carcinoma [
1]. The mechanisms related to viral replication and new virion assembly and release are important factors of HSV-2 infection. Following the entry of viral genomes into the nucleus, immediate early (IE), early (E), and late (L) genes lead to the generation of mature virions [
2].
HSV-2 infections are life-long, and the current standard treatment relies on acyclovir (ACV) and related synthetic nucleoside analogs that target the viral polymerase [
2]. However, these drugs cannot cure the infections; thus, reactivation can occur, particularly in immunocompromised individuals. Furthermore, long-term treatment with these drugs can lead to drug resistance [
3,
4]. To overcome this problem, several strategies have been proposed on other targets. Therefore, the use of natural products has gained much attention.
Natural products are a great source of several bioactive molecules and are usually used as alternative treatments due to their safety, therapeutic properties, and low cost [
3,
5]. Moreover, they have also proved to be a promising way to overcome challenges with drug resistance since their mode of action has been reported in various ways [
1]. Propolis is a natural resinous substance produced by bees that has been widely used as a traditional medicine for centuries [
6]. Numerous components have been identified in propolis, mainly polyphenol (flavonoids, phenolics, and esters), depending on the bee species, botanic and geographic origin [
7,
8]. Investigation of active ingredients in brown propolis was earlier performed by our group. The chromatogram profile analyzed by means of HPLC presented its chemical compositions: gallic acid, quercetin, pinocembrin, chrysin, and galangin [
9]. Moreover, propolis has been shown to display potent biological properties, such as antimicrobial, anti-inflammatory, anticancer, and antioxidant properties. Notably, its antiviral effect has also been proven [
5,
10]. The antiviral activity of propolis has been investigated against pathogenic human viruses such as influenza [
11], human immunodeficiency virus (HIV) [
12], human coronaviruses (SARS-CoV-2) [
5], and herpes simplex viruses (HSV-1 and HSV-2) [
13]. It was suggested that propolis inhibits HSV-2 infection through the destruction of the viral envelope and cellular absorption [
13,
14]. However, dissolubility of propolis has limited its therapeutic application and needs to be improved [
15].
In recent years, nanotechnology has been extensively applied in pharmaceutical and medical sciences by using nanoparticle-based drug delivery systems. These systems not only provide an efficient carrier, but also resolve the problem of dissolution of hydrophobic substances, resulting in an elevated level of therapeutic efficacy [
16,
17]. In our previous work conducted by Iadnut et al., 2019, a polymeric nanoparticle platform of a poly(lactic-co-glycolic acid) (PLGA) formulation was employed to encapsulate the ethanolic extract of propolis (EEP). This nanoformulation possessed a great characteristic of lower cytotoxicity, and the water solubility of EEP was also improved. However, a weak point associated with the zeta potential (ZP) of the nanoparticle surface was present and required a charge modification to stabilize its nanostructure [
9]. Modification of the surface charge can be rendered by the addition of negative or positive charge-inducing agents [
18]. In this study, chitosan (CS), a natural biopolymer, was used to produce cationic nanoparticles (NPs) that are known to have low toxicity with nonimmunogenic and biodegradable nature [
19,
20]. Besides an increase in the ZP, we also hypothesized that CS-modified PLGA NPs loaded with EEP could influence HSV-2 activity through the disruption of viral replication-related factors such as the IE or L genes.
Thus, this work was aimed at encapsulating EEP within CS-modified PLGA NPs (EEP-NPs) and characterizing their physicochemical properties, anti-HSV-2 activity, and mechanisms of actions.
3. Discussion
Natural product-based nanoparticles have been proposed as alternatives to treat infections and overcome the problem of drug resistance [
21,
22,
23]. We developed a nanoparticle-based drug delivery system encapsulating propolis and analyzed the anti-HSV-2 properties of these nanoparticles.
Propolis serves as a source of numerous active molecules with broad-spectrum antimicrobial activity, as well as antiviral properties. The effect of propolis against HSV-2 has been reported in many studies. It was demonstrated that propolis extracts from the bee glue of
Apis mellifera and Canadian propolis have a virucidal effect and interrupt HSV-2 activity in the virus adsorption step [
14,
24]. Yildirim et al., 2016, also observed the suppression of HSV-2 replication by Hatay propolis along with the combination with ACV, showing a synergetic antiviral activity that was superior to the treatment with ACV alone [
25]. Although propolis has proved its powerful activity, its poor solubilization has been noted [
15].
We previously showed that EEP can be successfully loaded within PLGA NPs to improve its water solubility; however, these nanoparticles were unstable due to the insufficient ZP value of the surface charge [
9]. The surface charge depends on the type of constituents used in the NPs. CS, a cationic biopolymer, is widely used to modify PLGA-based NPs via ionic crosslinking between the positive amino groups present in the CS chains and an oppositely charged agent [
26,
27]. Based on the previous formulation of PLGA NPs [
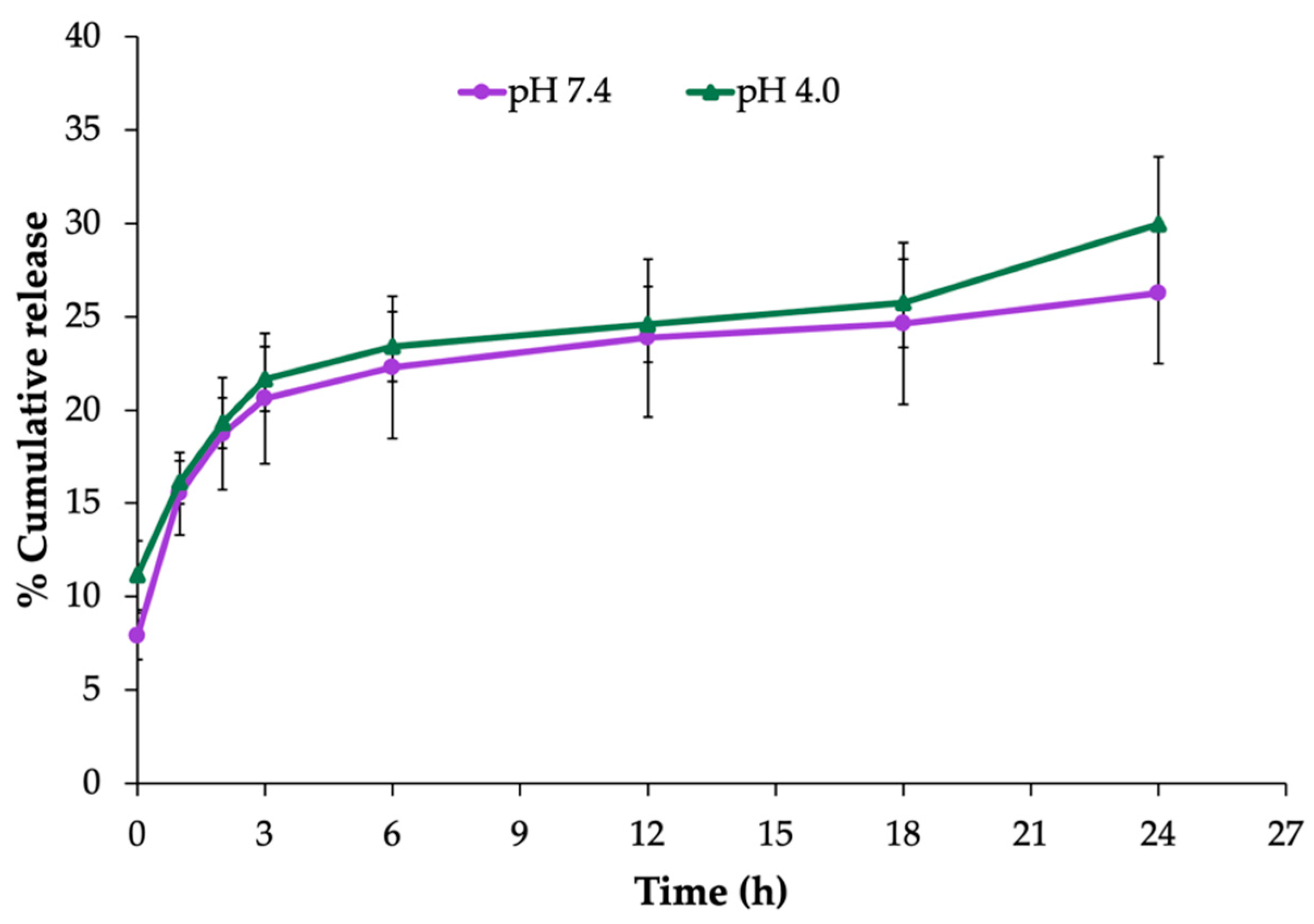
9], CS was added into the formulation to create a ZP of the NP surface. The modified formulation was improved as to be positively charged with the ZP value of approximately +38 mV resulting from the characteristics of CS and properly presented the average size at 450 nm and a PDI value of 0.21. In addition, these nanoparticles were able to release EEP in a sustained manner at pH 7.4 (stimulating the pH of a normal human body) and pH 4.0 (representing a vaginal pathological condition). In concordance with a previous study [
9], rapid release was observed within the first 3 h which lasted for up to 24 h. Nonetheless, the release rate was lower than what was reported. This difference in the release rate may have been due to the adsorption of drugs on CS through charge interaction which may influence the reduction of drug leakage [
27,
28].
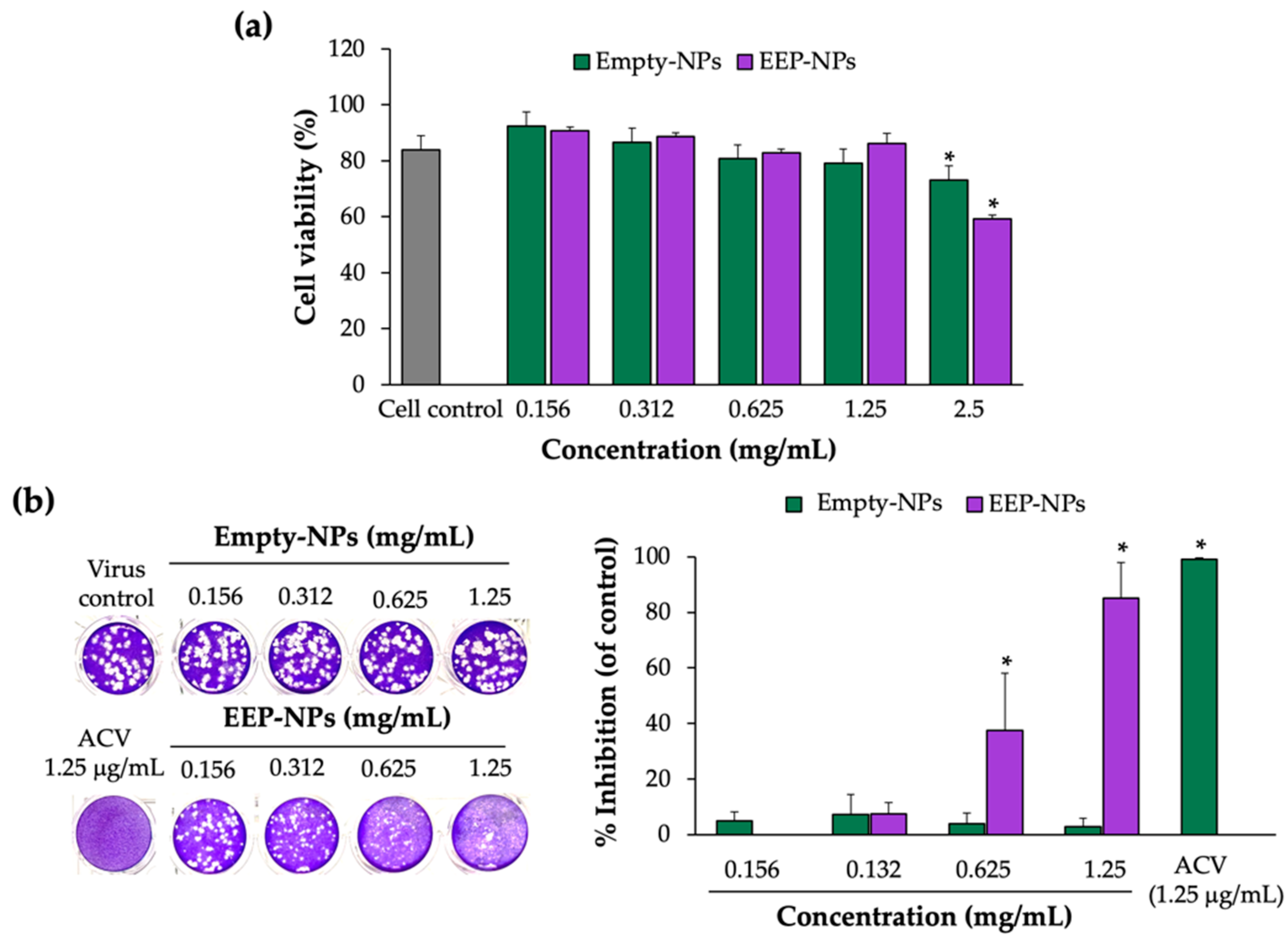
Since the biological study was performed, the cytotoxicity of the EEP-NPs was first examined. In concordance with the previous work, the EEP-NPs presented less toxicity to the Vero cells [
9]. These results indicated a suitable biomedical application of PLGA and CS as nanocarriers because of their nontoxic, biodegradable, and biocompatibility properties [
27]. Moreover, the safety of EEP was widely reported as well as in Vero cells [
29,
30].
Subsequently, low toxic concentrations of the EEP-NPs were tested for antiviral activity. Our results showed the antiviral effects of the EEP-NPs after treatment of the HSV-2-infected cells for 24 h in a dose-dependent manner. Moreover, the SI value also indicated nontoxicity of the EEP-NPs as previously described [
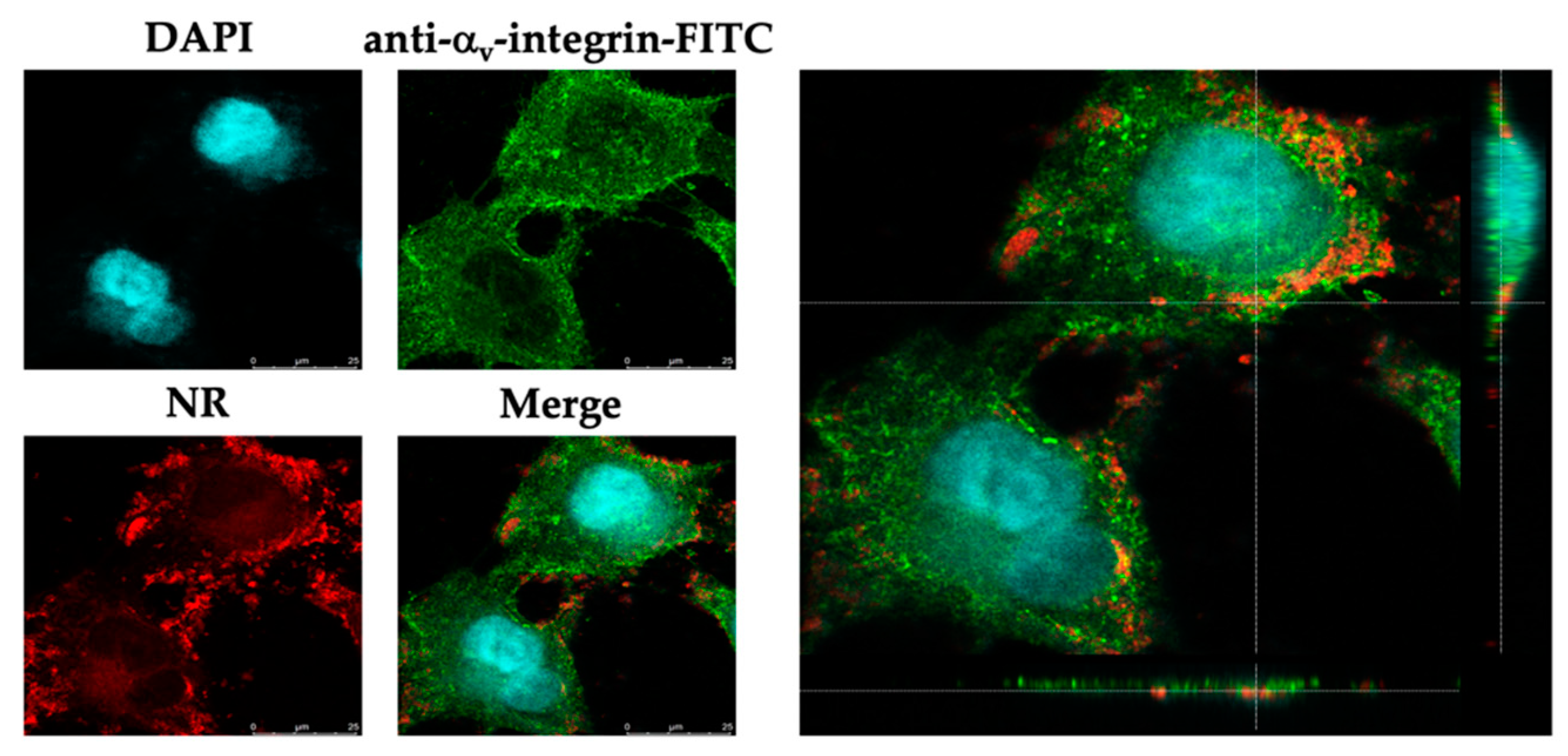
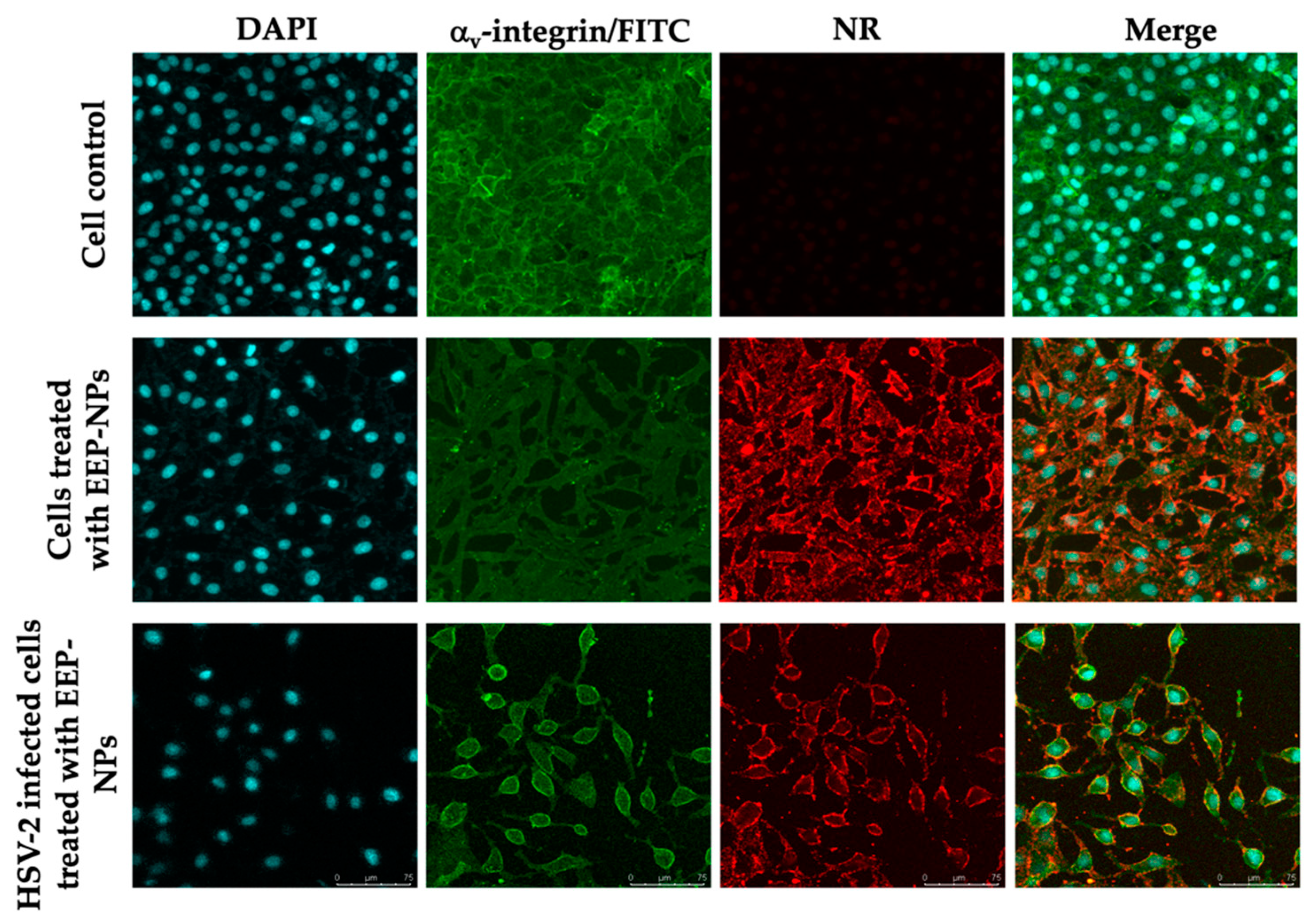
31]. However, many variations of acceptance criteria of SI values have been reported. Thus, further investigation is still required. Then, the CLSM analysis was conducted to show the uptake of the NPs into the infected cells (
Figure 4). The result demonstrated that the EEP-NPs could enter the infected cells and were located mainly in the cell membrane, followed by the cytoplasm.
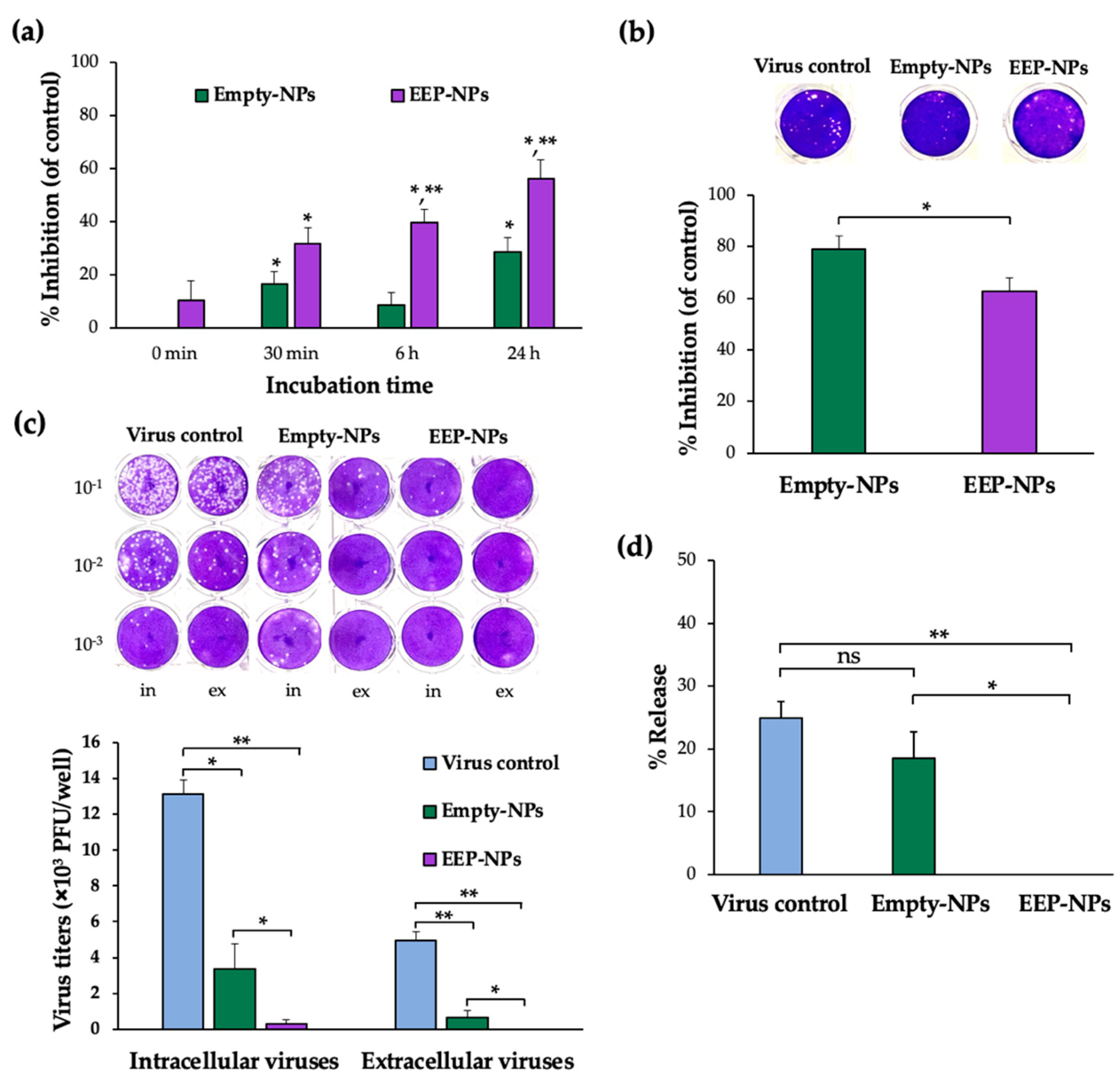
The role of the EEP-NPs against HSV-2 was further investigated by different modes of infection. The restriction of viral particles by the EEP-NPs was first elucidated. The significant direct inactivation effect on free HSV-2 by the EEP-NPs occurred within the first 30 min and up to 24 h of exposure. The Empty-NPs also inactivated HSV-2 virions, possibly due to the degradation of the NPs and the release of cationic CS.
Likewise, after the preincubation of the cells with the cationic NPs (EEP-NPs or Empty-NPs) with a negatively charged cell surface [
27], ultimately, the viral attachment, adsorption, and entry processes were blocked. Noticeably, the Empty-NPs showed a higher activity than the treatment with the EEP-NPs in this step. This effect was probably influenced by the dissociation of CS from the Empty-NPs, leading to ionic adsorption between CS and the cell surface, while incorporation of EEP in the formulation might result in the interference with or neutralization of the surface charge of CS; therefore, accumulation of CS on the cell surface may be reduced. Moreover, in the viral release assay, some intracellular and extracellular viruses were retained after treatment with the Empty-NPs. Noticeably, the EEP-NPs eliminated almost all of the intracellular and extracellular viruses as shown in
Figure 5c. These results indicated that both the EEP-NPs and the Empty-NPs disrupted the viral release affecting budding, or cell-to-cell spread of viruses. These assumptions were consistent with the intense fluorescence of the EEP-NPs mostly at the membrane (
Figure 6). This effect can be explained by the ionic adsorption between the characteristics of positively charged CS-based NPs and negatively charged cell membranes [
27,
32].
Our findings can be explained by two main aspects. Firstly, CS has an intrinsic antimicrobial property including antiviral activity [
23]. It has been proposed that the electrostatic interaction of positively charged CS and the negatively charged surface of the virus can inhibit the viral activity and/or directly kill the virus through viral protective membrane disruption [
23,
33]. Moreover, the viral capsid proteins or other virus-specific proteins could be interrupted by CS or its derivatives, leading to the prevention of viral glycoproteins and the interaction with their receptors, the reduction of viral entry, and eventually the suppression of viral replication [
23,
34,
35]. Secondly, the active constituents of EEP released from nanocarriers might directly affect the virion envelope, capsid proteins, or mask viral compounds such as glycoproteins, which are responsible for adsorption or entry into host cells, as described elsewhere [
14,
36]. It was stated that the constituents of propolis, galangin, and chrysin are responsible for antiviral activity by reducing the plaque formation of free HSV. However, the effect of propolis on virus replication was not detected [
37]. Thus, we hypothesized whether the EEP-NPs were involved in HSV-2 replication.
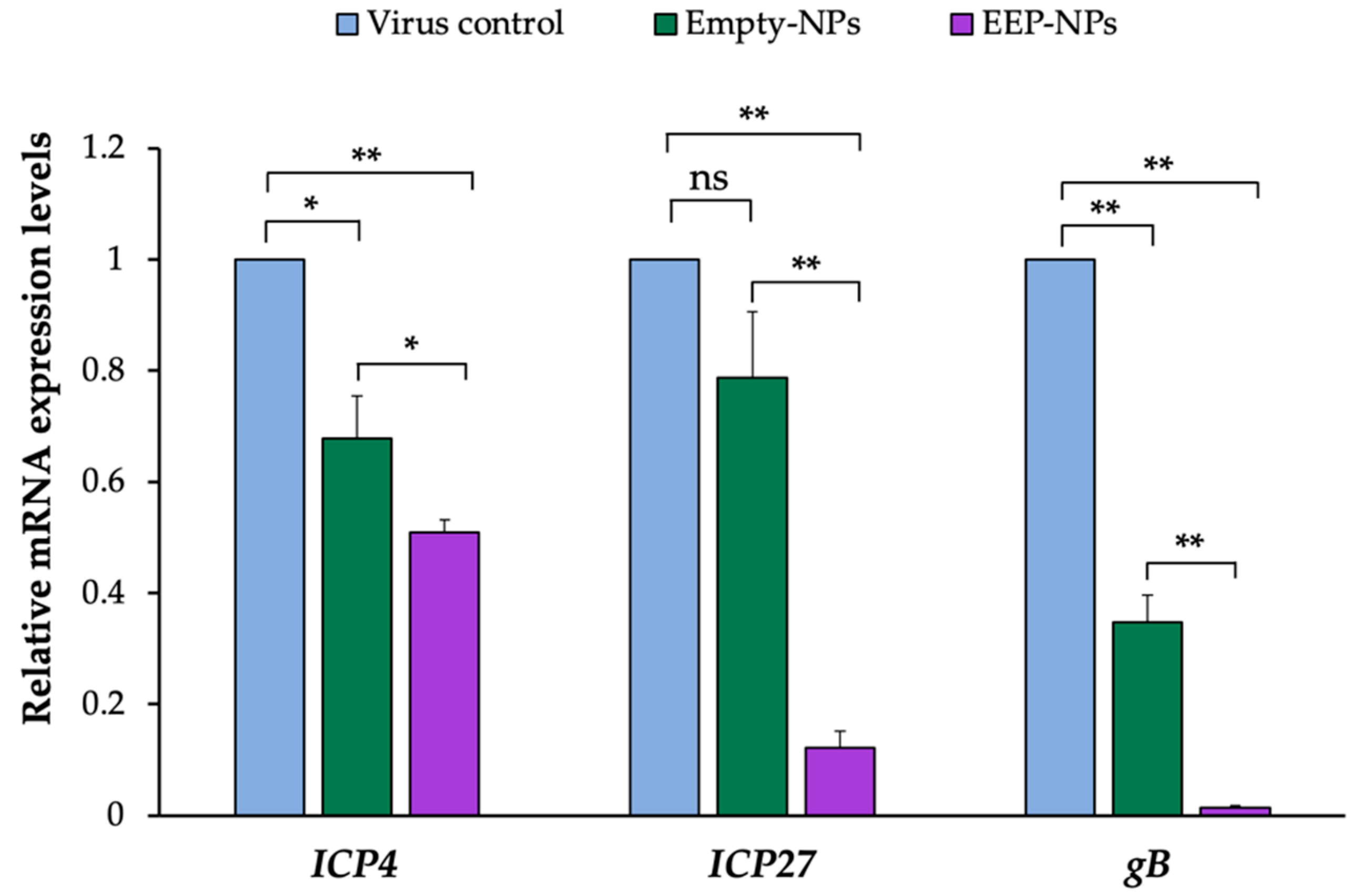
After entering the host cells, the viral genome is transported and released into the nucleus where the replication cycle takes place. HSV replication cascade is a sequential expression of the IE, E, and L gene products. Initially, the
IE genes are present (2–4 h of infection), and the IE proteins are required to trigger the transcription of the
E (4–9 h of infection) and
L genes (24 h of infection), respectively. Among the five viral IE regulatory proteins (ICP0, ICP4, ICP22, ICP27, and ICP47), ICP4 and ICP27 are phosphoproteins that are required to induce the expression of the
E and
L genes and are essential for HSV replication and infection [
38,
39,
40]. Moreover, late-stage protein gB was also chosen to be analyzed according to the low activity of viral release. Late-stage protein gB, a class III fusion glycoprotein, acts as a viral fusogen. The fusion reaction delivers the viral nucleocapsid and tegument proteins into the host cell [
41]. As shown in the result, after the administration of the EEP-NPs (or the Empty-NPs), the mRNA expression levels of the related factors,
ICP4,
ICP27 and
gB genes, were statistically decreased and the reduction of
L gene
gB was ascribed as the consequence of downregulation of
IE genes
ICP4 and
ICP27. This implied that the NPs were active in inhibiting HSV-2 replication. The suppression of HSV-2 replication might be caused by the cationic character of CS, enabling binding to negatively charged DNA or RNA [
42], thereby interfering with the viral genome. Notably, more inhibitory effects were found when the infected cells were applied to the NPs containing EEP. Regarding these results, the active EEP was able to present intracellular activity based on the nano-delivery system, thereby increasing the therapeutic potential of EEP. EEP might interfere not only with the superficial structure of the virion as mentioned above, but with the bioactive substances derived from EEP such as flavonoids, also probably inhibiting viral polymerase, binding to the viral nucleic acid, thus interrupting nucleic acid synthesis [
43,
44]. This suggests dual effects resulting from CS and EEP. Similarly, another work also introduced the use of CS in nanosystems to increase antiviral activity when compared to the drug alone [
23,
45].
Since this is the first time that the encapsulation of EEP in CS-modified PLGA NPs was investigated for application in HSV-2 treatment, more investigations regarding its characteristics and molecular mechanisms of antiviral activity need to be carried out. We also suggest that this nanoformulation could be further designed as a new topical formulation to inhibit viral replication that exists in the epidermis or the basal layer.
4. Materials and Methods
4.1. Propolis, Chemicals, and Reagents
The ethanolic extract of propolis (EEP) was kindly provided by Bee Products Industry (Lamphun, Thailand). Poly(lactic-co-glycolic acid) (PLGA) (lactide:glycolide = 50:50; inherent viscosity = 0.45–0.60 dL/g, Mw = 38–54 kDa) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Chitosan (CS) was kindly gifted by Assist. Prof. Dr. Worrapan Poomanee, Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University. Polyvinyl alcohol (PVA) and ethanol (EtOH) were purchased from Fluka (Buchs, Switzerland) and Merck Millipore (Darmstadt, Germany), respectively. Dichloromethane (DCM) was obtained from RCI Labscan (Gliwice, Poland). All the other chemicals and reagents used in this study were of analytical and molecular grade.
4.2. Preparation and Characterization of EEP-Loaded CS/PLGA Nanoparticles (EEP-NPs)
4.2.1. Preparation of EEP-NPs
Formulations of EEP-NPs (or Empty-NPs) were prepared using the modified oil-in-water (
o/
w) single emulsion solvent evaporation method with some modifications [
9]. Briefly, an organic solution consisting of 100 mg/mL EEP and 100 mg PLGA dissolved in DCM was prepared and added dropwise to an aqueous solution containing 0.4% (
w/
v) PVA and 1% (
w/
v) CS to obtain the ratio of 1:2 (
v/
v) of organic and aqueous phases. The resulting solution was stirred, and then sonicated using an ultrasonic processor UP50H (Hielscher Ultrasonics, Hielscher, NJ, USA) at 90% amplitude for 30 min within an ice bath. For complete polymerization, each mixture was stored overnight at room temperature in the dark. Afterwards, the solution was centrifuged at 8800×
g for 40 min at 4 °C to obtain NPs. The NPs were further washed once and reconstituted with deionized water before lyophilization. The NP samples were stored at −20 °C until used.
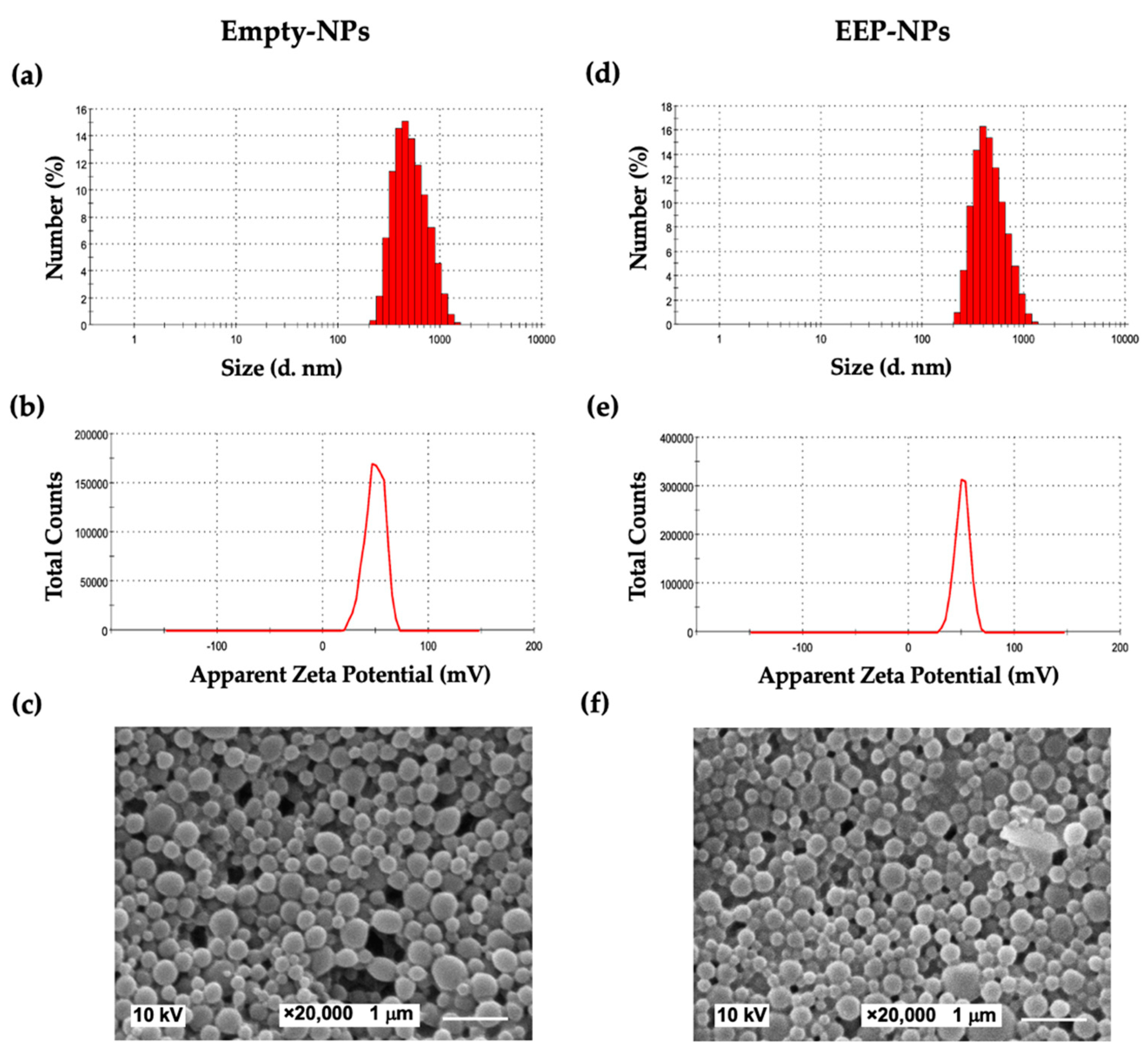
4.2.2. Physicochemical Characterization
The average particle size, polydispersity index (PDI) values, and zeta (ζ) potential (ZP) of the surface charge of the nanoformulations were evaluated using a Malvern Zetasizer Nano ZSP system (Malvern Instruments, Worcestershire, UK). The measurements were carried out for the EEP-NPs and the Empty-NPs at a 1/100 dilution in deionized water. Each sample was measured based on at least three measurements in three individual runs.
- 2.
Scanning electron microscopy (SEM)
The morphology of the NPs was observed using a scanning electron microscope (SEM) (JEOL, Tokyo, Japan). A drop of the formulation was air-dried on a copper tape and coated with a gold film using a JEOL JFC1100E Ion Sputter (JEOL, Tokyo, Japan) under vacuum for 2 min. After preparation, the shape and size of the particles were studied using SEM at an accelerating voltage of 10 kV.
- 3.
Encapsulation efficiency (EE) and loading capacity (LC)
The encapsulated EEP of the NPs was determined by means of an indirect quantification method. The supernatant of the EEP-NPs was obtained after centrifugation at 12,000×
g for 1 h at 4 °C. The supernatant was properly diluted in deionized water, and the absorbance was then measured at 290 nm using a UV–Vis spectrophotometer. The amount of EEP in the supernatant was determined using the standard calibration curve of EEP. The EE and LC of the EEP-NPs were quantified using the following Equations (1) and (2):
4.2.3. In Vitro Release Assay
The EEP release from the NPs was performed using a modified dissolution method [
40]. One milligram of the EEP-NPs was dissolved in 1 mL of a phosphate-buffered saline solution (PBS, pH 7.4 or 4.0) and shaken at 45 rpm in an incubator at 37 °C. The samples were collected at different timepoints (0, 1, 2, 3, 6, 12, 18, and 24 h) and centrifuged at 9800×
g for 20 min. The supernatant was harvested, and the pellet was resuspended with 1 mL of fresh PBS. The release of EEP from the NPs in the supernatant at the specified time interval was measured using a UV–Vis spectrophotometer at 290 nm. The percentage of EEP released from the NPs (% accumulative release) was compared with the EEP’s standard calibration curve.
4.3. Cell Culture and Virus
African green monkey kidney epithelium (Vero) cells (ATCC CCL-81) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (10 units/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2. The HSV-2 virus was obtained from Prof. Dr. Chalobon Yoosook, Department of Microbiology, Faculty of Science, Mahidol University. Acyclovir (Sigma-Aldrich, Darmstadt, Germany) was kindly gifted by Assist. Prof. Dr. Yingmanee Tragoolpua, Department of Biology, Faculty of Sciences, Chiang Mai University.
4.4. Cytotoxicity of EEP-NPs
The effect of the EEP-NPs and the Empty-NPs on the viability of the Vero cells was determined by means of the trypan blue assay. The cells (1 × 104 cells/well) were seeded in a 96-well plate. On the following day, different concentrations of the EEP-NPs (or the Empty-NPs) were added and incubated for 24 h. Then, the cells were harvested by trypsinization, and the number of live and dead cells was determined. The cytotoxicity of the NPs was expressed as the % cell viability, and the 50% cytotoxic concentration (CC50) was then calculated.
4.5. Assessment of Antiviral Activities
4.5.1. Determination of the Virus Titer Using a Plaque Assay
A plaque assay was performed according to Deethae et al., 2018 [
46], with some modifications. In brief, Vero cells (1 × 10
5 cells/well) were seeded in a 24-well plate overnight. The cell monolayer was infected with the HSV-2 virus at various dilutions and incubated at 37 °C in a 5% CO
2 incubator for 1 h. The infected cells were then overlaid with an overlay medium. After 48 h, the cells were washed three times with PBS and stained with 1% crystal violet solution. The plaques were counted, and plaque formation units (PFUs) were calculated.
4.5.2. Investigation of Antiviral Activity of the EEP-NPs against HSV-2 Infection
For the treatment assay [
46], the Vero cell monolayer was first incubated with 30 PFU/well of the HSV-2 virus at 37 °C for 1 h to allow viral attachment. Then, the EEP-NPs (or the Empty-NPs) with different concentrations were added to the infected cells, followed by an overlay medium. Quantification of the virus was performed using the plaque assay as described above (
Section 4.5.1). The infected cells without treatment with the EEP-NPs (or the Empty-NPs) were used as the virus control. The antiviral activity was determined by the percentage of HSV plaque inhibition (%) using the following Equation (3):
where V
C and V
T refer to the number of plaques in the absence and presence of EEP-NPs (or Empty-NPs).
Furthermore, the selectivity index (SI) was also calculated to indicate the toxicity of the EEP-NPs for normal cells as compared to viruses using the following Equation (4):
where CC50 and IC50 refer to the 50% cytotoxic concentrations and 50% inhibition concentrations, respectively.
4.5.3. Direct Inactivation Assays
To examine whether the EEP-NPs directly inhibited the viral particles, a direct inactivation assay was performed according to Deethae et al., 2018, with some modifications [
46]. Briefly, the HSV-2 virus supernatant at 200 PFU/mL was pretreated with the EEP-NPs (or the Empty-NPs) and kept at 4 °C for 0 min, 30 min, 6 h, and 24 h. Then, the treated viruses were added to the cells and incubated for another 48 h based on the plaque assay as described above (
Section 4.5.1)
4.5.4. Virus Release
The monolayer cell culture was infected with 30 PFU/well of the virus for 1 h to allow viral attachment. After 24 h treatment of the infected cells with the EEP-NPs (or the Empty-NPs), the supernatant and the cell pellet were collected, respectively, and the virus release was examined according to Wang et al., 2020 [
40], with some modifications. The cell pellet was subjected to three freeze–thaw cycles before titration. The plaque assay was performed, and virus titers of the supernatant and the cell pellet were determined. The virus release rate after treatment with the EEP-NPs (or the Empty-NPs) was also calculated using the following Equation (5):
where T
ex and T
in represent the extracellular and intracellular virus titers, respectively.
4.5.5. Expression Levels of Immediate Early (IE) and Late (L) HSV-2 Genes Using Quantitative Real-Time PCR
For the analysis of mRNA expression of the immediate early
ICP4 and
ICP27 and glycoprotein (
gB) HSV-2 genes, total RNA of the infected cells treated with the EEP-NPs (or the Empty-NPs) with a different post-infection (p.i.) time was extracted with the TRIzol reagent (Ambion, CA, USA) according to the manufacturer’s instructions and reverse-transcribed into cDNA using a Thermo Scientific RevertAid First Strand cDNA synthesis kit (Thermo Scientific, Waltham, MA, USA). The amplifications were carried out using a SensiFAST SYBR kit (BIOLINE, London, UK) using primers for
ICP4,
ICP27, and
gB. Glyceraldehyde-3-phosphate dehydrogenase (
GAPDH) was used as the internal control. The sequence of primers used in the PCR analysis is shown in
Table 3. The PCR reactions included initial denaturation at 95 °C for 120 s, followed by 40 cycles of denaturation at 95 °C for 10 s and annealing at 59 °C (
ICP4 and
ICP27) or 61 °C (
gB) for 31 s. The relative fold change in mRNA expression was analyzed using the 2
−ΔΔCT method. All the experiments were performed in three independent trials.
4.6. Fluorescence Staining Assay
The localization of the EEP-NPs was investigated using a confocal laser scanning microscope (CLSM). The cell monolayer was infected with HSV-2 and incubated at 37 °C, 5% CO2, for 1 h. To track the EEP-NPs, a 500 μL sample of the EEP-NPs was stained with 40 μL of 0.25 mg/mL Nile red (NR) solution for 20 min in the dark, at room temperature. After washing, the pellet was resuspended and subjected to treatment with the infected cells for 24 h. Then, the cells were fixed with 4% paraformaldehyde for 20 min, followed by a wash with PBS. Afterwards, the fixed cells were blocked with 2% bovine serum albumin at room temperature for 30 min and incubated with a mouse anti-αv-integrin mAb (kindly gifted by Prof. André Lieber, Department of Medicine, University of Washington, Seattle, WA, USA). After being washed, the cells were incubated with an FITC-conjugated rabbit anti-mouse polyclonal antibody (Dako, Santa Clara, CA, USA) (kindly gifted by Assoc. Prof. Dr. Sawitree Chiampanichayakul, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University). The cell nuclei were then counterstained with 4′,6-diamidio-2-phenylindole (DAPI) (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Images were acquired using a confocal laser scanning microscope (Leica TCS SP8, Mannheim, Germany).
4.7. Statistical Analysis
The results are expressed as the means ± SEM. All the experiments were repeated in three independent trials. Comparisons between the groups were performed using a one-way analysis of variance and post hoc LSD. Significant differences were indicated by p < 0.05 or 0.001. All the calculations were performed using the SPSS Statistics 23.0 software (IBM Corp., Armonk, NY, USA).


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}