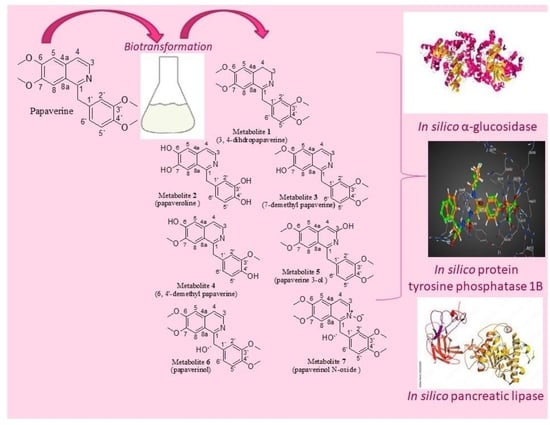
2.1. Identification of the Biotransformation Products
Fifty different types of fungi of different classes were used in the initial screening of isoquinoline alkaloid substrate papaverine. Seven biotransformation products of papaverine were isolated using different fungi: Cunninghamella elegans NRRL 2310, Penicillium chrysogeneum ATCC 10002, Rhodotorula rubra NRRL y1592 and Cunninghamella blackesleeana NRRL 1369. Structural elucidation of metabolites was based on their mass and NMR spectroscopic data.
The mass spectrum of
1 (SI: S5) displayed a molecular ion peak at
m/
z 341, corresponding to a molecular formula of C
20H
23NO
4, which is 2 Da higher than that of papaverine. Peaks at
m/
z 326, 310, 309 and 295 corresponded to (M-CH
3)
+, (M-OCH
3)
+, (M-H-OCH
3)
+ and (M-OCH
3-CH
3)
+ fragments, respectively. When compared with papaverine,
1H NMR (
Table 1, SI:S1) displayed the absence of doublets of one-proton intensity each at δ
H 8.33 ppm (J = 5.6 Hz) and 7.53 ppm (J = 5.6 Hz), belonging to two protons, H-3 and H-4, respectively, that are present in papaverine. It also displayed the appearance of two other triplets of two protons’ intensity at δ
H 3.47 (J = 7.2 Hz) ppm and 3.07 (J = 7.2 Hz) ppm, assigned to four protons on saturated carbons at H-3 and H-4, respectively, which suggested a reduction in C-3 and C-4 positions. A total of twenty-three protons resonating between δ
H 3.07 and 7.38 ppm compared to twenty-one protons in papaverine confirmed reduction in the dihydro derivative. In
1,
13C NMR (
Table 2, SI: S2) showed twenty signals corresponding to twenty different carbons and the disappearance of two carbon signals, at δ
C 119.6 and 139.0 ppm, present in papaverine. Furthermore, two high field resonances, at δ
C 46.5 ppm (C-3) and 26.2 ppm (C-4), substantiated the reduction of papaverine. Other
13C NMR data exhibited resemblance to those of papaverine. Carbon multiplicity experiments (DEPT-135 and APT spectra) on metabolite
1 (SI: S3, SI: S4) proved the existence of three “methylene” carbons (attributed to C-α, C-3 and C-4), four “methyl” carbons (C-3′, C-4′, C-7, C-6) and five “methine” carbons (C-5, C-8, C-6′, C-5′, C-2′). The IR data (SI: S6) showed a characteristic broad band of N–H stretching at 3430 cm
−1, one of aliphatic C-C stretching at 1603 cm
−1 and one of tertiary alcoholic C–O stretching at 1157 cm
−1. All information matched that which had been reported for 3, 4-dihydropapaverine [
20] (
Figure 1).
The mass spectrum of
2 (SI: S11) exhibited a molecular ion peak at
m/z 283, corresponding to a molecular formula of C
16H
13NO
4, which is 56 Da lower than that of papaverine. The
1H NMR data of
2 (SI: S7) compared to papaverine (
Table 1) showed the disappearance of three sharp proton singlets of 7, 6, 3′ and 4′ methoxy moieties, resonating at δ
H 4.02, 3.93, 3.78 and 3.80 ppm, respectively, and the appearance of another four broad singlets that disappeared upon addition of D
2O, integrating for four protons at δ
H 5.06, 5.15, 5.33 and 5.64 ppm, due to four OH groups resulting from the demethylation reaction at C-6, C-7, C-3′ and C-4′, respectively. Of
2,
13C NMR (
Table 2 and SI: S8) showed sixteen signals, which corresponded to sixteen different carbon atoms reminiscent to those of papaverine, with the sole difference being the lack of four methoxy carbon signals of papaverine at δ
C 55.9, 56.12, 56.10 and 56.4 ppm. Carbon multiplicity experiments (DEPT-135 and APT) on
2 (SI: S9, SI: S10) confirmed the presence of one “methylene” carbon (C- α) and seven “methine” carbons (C-3, C-4, C-5, C-8, C-6′, C-5′ and C-2′). The IR spectrum (SI: S12) of
2 exhibited a hydroxy group band at 3455 cm
−1, which was missing in the papaverine spectrum as reported (SI: S12). All data were coincident with those published for papaveroline [
21] (
Figure 1).
The mass spectrum of
3 (SI: S19) exhibited a molecular ion peak at
m/
z 325, corresponding to a molecular formula of C
19H
19NO
4, which is 14 Da lower than that of papaverine. Peaks at
m/
z 324, 310, 294, 293 and 279 corresponded to (M-H)
+, (M-CH
3)
+, (M-OCH
3)
+, (M-H-OCH
3)
+ and (M-OCH
3-CH
3)
+ fragments, respectively. The presence of fragments at
m/
z 188 and 151 increased the evidence of a C-7 demethylation reaction, as previously reported. The IR spectrum (SI: S20) of
3 showed an OH group band at 3438 cm
−1, which was missing in the papaverine spectrum. The
1H NMR spectra of 3 (
Table 1, SI: S13) lacked a singlet signal at δ
H 3.93 ppm, integrating for three protons (C-7 methyl group) present in papaverine, and the appearance of another broad singlet, integrating for one proton at δ
H 6.02 ppm, due to the OH group resulting from the demethylation reaction. For
13C NMR (
Table 2, SI: S14), carbon multiplicity experiments (DEPT-135 and APT spectra) (SI: S15 and SI: S16) also showed the disappearance of a signal at δ
C 56.10 ppm due to the C-7 methoxy group in papaverine. This was substantiated through extensive analysis of the HMBC and HSQC spectra of
3 (SI: S17, SI: S18). Based on the aforementioned spectral data, which were consistent with published data, metabolite
3 was identified as 7-demethyl papaverine [
11] (
Figure 1).
The mass spectrum (SI: S27) of
4 displayed a molecular ion peak at
m/
z 311, corresponding to a molecular formula of C
18H
17NO
4, which is 28 Da lower than that of papaverine. Peaks at
m/
z 310, 296, 280, 279 and 265 corresponded to (M-H)
+, (M-CH
3)
+, (M-OCH
3)
+, (M-H-OCH
3)
+ and (M-OCH
3-CH
3)
+ fragments, respectively. The presence of fragments at
m/
z 188 and 137 increased the evidence of C-6 and C-4′ demethylation reactions, as previously reported [
5]. In addition,
4, when compared to the papaverine, displayed a lack of 6 and 4`-methoxy groups resonating at δ
H 4.02 and 3.80 ppm in
1H NMR (
Table 1, SI: S21). For
13C NMR, carbon multiplicity experiments (DEPT-135 and APT spectra) on
4 (
Table 2, SI: S22, SI: S23 and SI: S24) showed two methoxy signals at δ
C 55.3 and 55.5 ppm, which are less than that of papaverine by two methoxy groups. This was substantiated with extensive analysis of the HMBC and HSQC spectra of
4 (SI: S25 and SI: S26). The IR spectrum (SI: S28) also revealed the presence of a hydroxy band at 3450 cm
−1, which is absent in the papaverine spectrum, as previously published. Hence,
4 was identified as 6, 4′-didemethyl papaverine [
11] (
Figure 1).
The
1H NMR spectrum of metabolite
5 compared to that of papaverine (
Table 1, SI: S29) displayed a lack of doublets of one-proton intensity each at δ
H 8.33 (J = 5.6 Hz) ppm and 7.53 (J = 5.6 Hz), belonging to two protons on H-3 and H-4, respectively, that are present in papaverine. It also displayed the appearance of another two singlets of one-proton intensity each, at δ
H 6.99 and 2.36 ppm, which belonged to one proton on C-4 and the OH group on C-3, respectively. The later signal disappeared on deuteration, which suggested hydroxylation of C-3. Examination of the
13C NMR of
5 (
Table 2, SI: S30) showed twenty peaks, which were related to twenty diverse carbon atoms. There was a considerable deshielding of C-3 carbon signals (by 25 ppm) and a considerable shielding of C-4 (by 20 ppm). Other
13C NMR data revealed resemblance to those of papaverine. The DEPT-135 spectra of
5 (SI: S31) confirmed the presence of one methylene carbon (assigned to C-α), four methyl carbons (C-3′, C-4′, C-7, C-6) and six methine carbons (C-5, C-8, C-6′, C-5′, C-2′, C-4). The mass spectrum (SI: S32) showed the presence of a molecular ion peak at
m/
z 355, presumably relating to a molecular formula of C
20H
21NO
5, which is 16 Da higher than that of papaverine. The IR spectrum (SI: S33) showed an OH band at 3432 cm
−1, which is not present in the papaverine spectrum, as previously reported. Peaks at
m/
z 354, 340, 325 and 309 corresponded to (M-H)
+, (M-CH
3)
+, (M-OCH
3)
+ and (M-OCH
3-CH
3)
+ fragments, respectively. Based on these data, metabolite
5 was elucidated as papaverine 3-ol [
22] (
Figure 1).
The positive ESI-MS spectrum (SI: S38) of
6 exhibited [M+H]
+ at
m/
z 356 of a pseudomolecular ion peak, apparently indicating a molecular formula of C
20H
22O
5N, which is 16 Da higher than that of papaverine (one extra oxygen atom). The
1H NMR data of
6 (
Table 1, SI: S34) showed that the metabolite spectrum exhibited four sharp three-proton singlets at δ
H 3.95, 3.71, 3.76 and 3.81 ppm, assigned to the 6, 7, 3′ and 4′ methoxy groups, respectively. It also lacked a singlet signal of two protons’ intensity at δ
H 4.56 ppm, belonging to two protons on carbon α, which is present in papaverine, and the presence of a broad singlet at δ
H 6.08 ppm, integrating for one proton. The remaining chemical shifts of proton signals were very close to those of papaverine, as reported. The
13C NMR of
6 (
Table 2, SI: S35) showed twenty peaks, which related to twenty various carbon atoms similar to the total number of carbon atoms of papaverine, but there was considerable deshielding of C-α (by 30 ppm) and C-1′ (by 3 ppm). Both the DEPT-135 and APT spectra of
6 (SI: S36, SI: S37) exhibited the absence of one methylene carbon (attributed to C-α) present in papaverine and the appearance of a new methine carbon at δ
C 72.5 ppm. In addition, seven other methine carbons (C-3, C-4, C-5, C-8, C-2′, C-5′, C-6′,) and four methyl carbons (C-3′, C-4′, C-6, C-7) were observed. The IR spectrum (SI: S39) showed a strong absorption band at 3345 cm
−1, suggesting the presence of hydroxy functionality, which is lacking in the papaverine spectrum, as formerly reported. Based on
6’s spectral data, it was identified as papaverinol that was previously isolated from microbial transformation of papaveraldine via Mucor ramannianus 1839 [
10], However, this is the first report of papaverinol being isolated from biotransformation of papaverine (
Figure 1).
The (+) ESI-MS spectrum (SI: S44) of
7 displayed a pseudomolecular ion peak [M + H]
+ at
m/
z 372, indicating a molecular formula of C
20H
22O
6N, which is 32 Da higher than that of papaverine (two additional oxygen atoms). The
1H NMR data of
7 (
Table 1, SI: S40) displayed a lack of a singlet of two protons’ intensity at δ
H 4.56 ppm belonging to two protons on carbon α, which is present in papaverine, and the appearance of a broad singlet at δ
H 6.50 ppm, integrating for one proton, which disappeared upon addition of D
2O, in addition to a singlet signal at δ
H 6.1 ppm due to C-α. There was a slight deshielding in doublets of one-proton intensity at δ
H 8.41 (J = 5.2 Hz) ppm and δ
H 7.51 ppm (J = 5.2 Hz), belonging to two protons on unsaturated carbons and assigned to H-3 and H-4, respectively. Signals observed at δ
H 7.06 (1 H, s) ppm and δ
H 7.12 (1 H, s) were assigned to H-5 and H-8, respectively. Examination of the
13C NMR of
7 (
Table 2, SI: S41) showed twenty peaks, which corresponded to twenty various carbon atoms, similar to the total number of carbon atoms of papaverine, but there was considerable deshielding of C-α (by 30 ppm), C-1′ (by 4 ppm), C-1 (by 3 ppm) and C-3 (by 2 ppm). In carbon multiplicity NMR experiments, the DEPT-135 and APT spectra of
7 (SI: S42, SI: S43) showed the absence of one methylene carbon (attributed to C-α) present in papaverine and the appearance of a new methine carbon at δ
C72.5 ppm. In addition, seven other methine carbons (C-3, C-4, C-5, C-8, C-6′, C-5′, C-2′) and four methyl carbons (C-3′, C-4′, C-6, C-7) were present. The IR spectrum (SI: S45) showed a strong absorption band at 3437 cm
−1, suggesting the presence of hydroxy functionality, which is lacking in the papaverine spectrum, as formerly reported. Based on
7’s spectral data, it was identified as papaverinol N-oxide that was previously isolated through microbial transformation of papaveraldine via Mucor ramannianus 1839 [
10]; however, this is the first report of papaverinol N-oxide being isolated from biotransformation of papaverine (
Figure 1).
Most of the biotransformation reactions were observed to take place in the heterocyclic ring, reinforcing the binding of the aromatic ring to the active site of microbial enzymes and bringing the heterocyclic ring to direct contact with the modification site. Furthermore, the oxidative transformation of papaverine via hydroxylation, N-oxidation and dealkylation can be proposed to be mediated with cytochrome P450 monoxygenases [
23].
2.2. ADMET/Pharmacokinetic Properties and Drug-Likeness Predictions of Papaverine and Its Metabolites (1–7)
Through applying the necessary procedures in drug design, development and discovery endeavors, it is crucial to analyze several vital pharmacokinetic parameters, or ADMET qualities (absorption, distribution, metabolism, excretion and toxicity), as the most relevant attributes [
24,
25,
26].
The physicochemical characteristics of papaverine and its metabolites (
1–
7) are discussed in
Table 3 and
Figure 2A.
As shown in
Table 3, the papaverine and its metabolites met the necessities of drug likeness and passed their filters, such as the Veber filter (rotatable bonds ≤ 10 with TPSA ≤ 140) [
27,
28]. Furthermore, the papaverine and its metabolites were also verified with Lipinski’s rule of five (MW ≤ 500, HBA ≤ 10, HBD ≤ 5 and Log Po/w ≤ 5) [
29].
As shown in
Figure 2B–I, the papaverine and its metabolites (
1–
7) were investigated and divided into six sections with adequate ranges for oral bioavailability. These characteristics included lipophilicity, insolubility, size, insaturation, polarity and flexibility [
30]. The papaverine and its metabolites’ results were within these limits and had good physiochemical profiles, which is one of the factors that has to be observed in pharmaceuticals and clinical investigations (metabolites
2–
4 were exceptions because of insaturation).
Before a biomolecule is used in pharmaceutical or clinical trial fields for pharmaceutical formulation, it is crucial to have its HIA and CNS absorption evaluated [
31]. Blood–brain barrier penetration is crucial because it ensures that only substances that act on the central nervous system (CNS) can pass through it and that substances that have no effect on the CNS should not interact [
32]. The BOILED- Egg curve can be used to forecast drugs’ GI absorption (HIA) and BBB penetration [
33]. There are two areas: one for BBB penetration (yolk) and another for the GI absorption zone (HIA). Any component identified in the gray zone is not indicative of GI absorption or BBB penetration. The papaverine and all of its metabolites (apart from metabolite
2) showed high gastrointestinal absorption (HIA) and high BBB permeability, as shown in
Table 3. However, papaverine and other BBB-penetrating metabolites (apart from metabolite
1) are vulnerable to P-efflux gp’s mechanism.
In skin penetration according to previous reported research [
34], the log K
p values of papaverine and its metabolites were acceptable. The less permeant a molecule is in the skin, the bigger the negative value of K
p. Another advantage is that it anticipates the five main cytochrome (CYP) isoforms. These enzyme isoforms handle the metabolisms of nearly 75% of the drugs sold today and play a critical role in excretion of pharmaceuticals. Inhibiting any of these isoforms results in significant drug–drug interactions [
35]. Metabolites
2 and
7 did not block any cytochrome isoform (except CYP1A2), as shown in
Table 4, and were rapidly metabolized. Drug–medication interactions may occur when three or four cytochrome isoforms are inhibited by remaining metabolites. Drug clearance, which is calculated via summing the excretion rates from the liver and the kidney, determines the dosage rates needed to achieve steady-state concentrations. Furthermore, the clearance value for metabolite
7 was adequate. The unfavorable interactions that arise when organic cation transporter 2 (OCT2) inhibitors and substrates are combined may be influenced with OCT2 intermediates. It has been proposed that papaverine and all of its metabolites would serve as OCT2 nonsubstrates.
Before a medication enters the clinical trial stage or the production phase of the pharmaceutical industry, it is crucial to assess its toxicological profile [
36]. Each compound in this study had its many toxicities, including those that could harm the environment and people, evaluated (
Table 5). The mutagenic potential of a substance can be determined using the Ames test. Results showed that papaverine and all its metabolites were classed as non-Ames-hazardous, i.e., doubtful to be carcinogenic. It is likely that blocking the potassium channels that hERG encodes would result in dangerous ventricular arrhythmia. All examined compounds can suppress hERG II but not hERG I, according to numerous studies [
37]. However, it was anticipated that papaverine and metabolites
3–
5 would be hepatotoxic and cause liver damage as a result of the drugs. It was projected that metabolites
1–
2 and
6–
7 would not be hepatotoxic. Dermally applied items may produce skin hypersensitivity, although none of the compounds have been proven to make people’s skin more sensitive.
Additionally, compound metabolites
1 and
2 each received a score greater than 300 mg/kg for prediction of lethal doses (LD50) and were classified as class IV; as a result, they are considered “harmful if swallowed” (300 LD50 2000). Meanwhile, papaverine and metabolites
3–
7 had scores of over 2000 and were classified as class V; as a result, they are considered “may be harmful if swallowed” (2000) (
Table 5). According to their toxicological characteristics, papaverine and all of its metabolites are categorized as being in classes IV and V because they are not thought to pose a concern of protein poisoning.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}