

Synthesis and Cytotoxic Activity of the Derivatives of N-(Purin-6-yl)aminopolymethylene Carboxylic Acids and Related Compounds
 , , , and
, , , and
Abstract
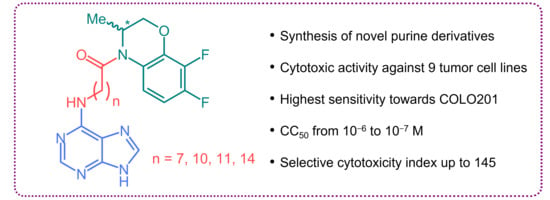
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Cytotoxicity Assessment
2.3. Cell Cycle Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. Chemistry General Section
3.1.2. Synthesis
General Procedure for the Synthesis of N-(Purin-6-yl)amino Carboxylic Acids 2a–c
3.2. Cytotoxicity Assessment
3.2.1. Materials
3.2.2. Cell Lines
3.2.3. MTT Cytotoxicity Assay
3.2.4. Cell Cycle Analysis
3.2.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Gray, N.S.; Rosania, G.R.; Sutherlin, D.P.; Kwon, S.; Norman, T.C.; Sarohia, R.; Leost, M.; Meijer, L.; Schultz, P.G. Synthesis and application of functionally diverse 2,6,9-trisubstituted purine libraries as CDK inhibitors. Chem. Biol. 1999, 6, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, L.; Raymond, E. Roscovitine and Other Purines as Kinase Inhibitors. From Starfish Oocytes to Clinical Trials. Acc. Chem. Res. 2003, 36, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Gucký, T.; Jorda, R.; Zatloukal, M.; Bazgier, V.; Berka, K.; Řezníčková, E.; Béres, T.; Strnad, M.; Kryštof, V. A Novel Series of Highly Potent 2,6,9-Trisubstituted Purine Cyclin-Dependent Kinase Inhibitors. J. Med. Chem. 2013, 56, 6234–6247. [Google Scholar] [CrossRef]
- Khalil, H.S.; Mitev, V.; Vlaykova, T.; Cavicchi, L.; Zhelev, N. Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J. Biotechnol. 2015, 202, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Gavilán, M.; Conejo-García, A.; Cruz-López, O.; Núñez, M.C.; Choquesillo-Lazarte, D.; González-Pérez, J.M.; Rodríguez-Serrano, F.; Marchal, J.A.; Aránega, A.; Gallo, M.A.; et al. Synthesis and Anticancer Activity of (R,S)-9-(2,3-Dihydro-1,4-Benzoxathiin-3-ylmethyl)-9H-Purines. ChemMedChem 2008, 3, 127–135. [Google Scholar] [CrossRef]
- Conejo-García, A.; García-Rubiño, M.E.; Marchal, J.A.; Núñez, M.C.; Ramírez, A.; Cimino, S.; García, M.Á.; Aránega, A.; Gallo, M.A.; Campos, J.M. Synthesis and anticancer activity of (RS)-9-(2,3-dihydro-1,4-benzoxaheteroin-2-ylmethyl)-9H-purines. Eur. J. Med. Chem. 2011, 46, 3795–3801. [Google Scholar] [CrossRef]
- Caba, O.; Díaz-Gavilán, M.; Rodríguez-Serrano, F.; Boulaiz, H.; Aránega, A.; Gallo, M.A.; Marchal, J.A.; Campos, J.M. Anticancer activity and cDNA microarray studies of a (RS)-1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl]-6-chloro-9H-purine, and an acyclic (RS)-O,N-acetalic 6-chloro-7H-purine. Eur. J. Med. Chem. 2011, 46, 3802–3809. [Google Scholar] [CrossRef]
- Knies, C.; Hammerbacher, K.; Bonaterra, G.A.; Kinscherf, R.; Rosemeyer, H. Nucleolipids of Canonical Purine β-D-Ribo-Nucleosides: Synthesis and Cytostatic/Cytotoxic Activities Toward Human and Rat Glioblastoma Cells. ChemistryOpen 2016, 5, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Salas, C.O.; Zarate, A.M.; Kryštof, V.; Mella, J.; Faundez, M.; Brea, J.; Loza, M.I.; Brito, I.; Hendrychová, D.; Jorda, R.; et al. Promising 2,6,9-Trisubstituted Purine Derivatives for Anticancer Compounds: Synthesis, 3D-QSAR, and Preliminary Biological Assays. Int. J. Mol. Sci. 2020, 21, 161. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.V.; Hoarau, C.; Bursavich, M.; Slattum, P.; Gerrish, D.; Yager, K.; Saunders, M.; Shenderovich, M.; Roth, B.L.; McKinnon, R.; et al. Lead optimization of purine based orally bioavailable Mps1 (TTK) inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4377–4385. [Google Scholar] [CrossRef]
- De las Infantas, M.J.P.; Torres-Rusillo, S.; Unciti-Broceta, J.D.; Fernandez-Rubio, P.; Luque-Gonzalez, M.A.; Gallo, M.A.; Unciti-Broceta, A.; Molina, I.J.; Diaz-Mochon, J.J. Synthesis of 6,8,9 poly-substituted purine analogue libraries as pro-apoptotic inducers of human leukemic lymphocytes and DAPK-1 inhibitors. Org. Biomol. Chem. 2015, 13, 5224–5234. [Google Scholar] [CrossRef] [Green Version]
- Bosco, B.; Defant, A.; Messina, A.; Incitti, T.; Sighel, D.; Bozza, A.; Ciribilli, Y.; Inga, A.; Casarosa, S.; Mancini, I. Synthesis of 2,6-Diamino-Substituted Purine Derivatives and Evaluation of Cell Cycle Arrest in Breast and Colorectal Cancer Cells. Molecules 2018, 23, 1996. [Google Scholar] [CrossRef] [Green Version]
- Cancilla, M.T.; He, M.M.; Viswanathan, N.; Simmons, R.L.; Taylor, M.; Fung, A.D.; Cao, K.; Erlanson, D.A. Discovery of an Aurora kinase inhibitor through site-specific dynamic combinatorial chemistry. Bioorg. Med. Chem. Lett. 2008, 18, 3978–3981. [Google Scholar] [CrossRef]
- Enkvist, E.; Lavogina, D.; Raidaru, G.; Vaasa, A.; Viil, I.; Lust, M.; Viht, K.; Uri, A. Conjugation of Adenosine and Hexa-(D-arginine) Leads to a Nanomolar Bisubstrate-Analog Inhibitor of Basophilic Protein Kinases. J. Med. Chem. 2009, 52, 308–321. [Google Scholar] [CrossRef]
- Rubio-Ruíz, B.; Conejo-García, A.; Ríos-Marco, P.; Carrasco-Jiménez, M.P.; Segovia, J.; Marco, C.; Gallo, M.A.; Espinosa, A.; Entrena, A. Design, synthesis, theoretical calculations and biological evaluation of new non-symmetrical choline kinase inhibitors. Eur. J. Med. Chem. 2012, 50, 154–162. [Google Scholar] [CrossRef]
- Liu, R.; Wang, J.; Tang, W.; Fang, H. Design and synthesis of a new generation of substituted purine hydroxamate analogs as histone deacetylase inhibitors. Bioorg. Med. Chem. 2016, 24, 1446–1454. [Google Scholar] [CrossRef]
- Karellas, P.; McNaughton, M.; Bake, S.P.; Scammells, P.J. Synthesis of Bivalent β2-Adrenergic and Adenosine A1 Receptor Ligands. J. Med. Chem. 2008, 51, 6128–6137. [Google Scholar] [CrossRef]
- Barlow, N.; Baker, S.P.; Scammells, P.J. Effect of Linker Length and Composition on Heterobivalent Ligand-Mediated Receptor Cross-Talk between the A1 Adenosine and β2 Adrenergic Receptors. ChemMedChem 2013, 8, 2036–2046. [Google Scholar] [CrossRef]
- Januchta, W.; Serocki, M.; Dzierzbicka, K.; Cholewinski, G.; Gensicka, M.; Skladanowski, A. Synthesis and biological evaluation of novel analogues of batracylin with synthetic amino acids and adenosine: An unexpected effect on centromere segregation in tumor cells through a dual inhibition of topoisomerase IIα and Aurora B. RSC Adv. 2016, 6, 42794–42806. [Google Scholar] [CrossRef] [Green Version]
- Gruzdev, D.A.; Musiyak, V.V.; Chulakov, E.N.; Levit, G.L.; Krasnov, V.P. Synthesis of purine and 2-aminopurine conjugates bearing the fragments of heterocyclic amines at position 6. Chem. Heterocycl. Compd. 2015, 51, 738–744. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Gruzdev, D.A.; Chulakov, E.N.; Vigorov, A.Y.; Musiyak, V.V.; Matveeva, T.V.; Tumashov, A.A.; Levit, G.L.; Charushin, V.N. Synthesis of novel purin-6-yl conjugates with heterocyclic amines linked via 6-aminohexanoyl fragment. Mendeleev Commun. 2015, 25, 412–414. [Google Scholar] [CrossRef]
- Eletskaya, B.Z.; Konstantinova, I.D.; Paramonov, A.S.; Esipov, R.S.; Gruzdev, D.A.; Vigorov, A.Y.; Levit, G.L.; Miroshnikov, A.I.; Krasnov, V.P.; Charushin, V.N. Chemoenzymatic arabinosylation of 2-aminopurines bearing the chiral fragment of 7,8-difluoro-3-methyl-3,4-dihydro-2H-[1,4]benzoxazines. Mendeleev Commun. 2016, 26, 6–8. [Google Scholar] [CrossRef]
- Eletskaya, B.Z.; Gruzdev, D.A.; Krasnov, V.P.; Levit, G.L.; Kostromina, M.A.; Paramonov, A.S.; Kayushin, A.L.; Muzyka, I.S.; Muravyova, T.I.; Esipov, R.S.; et al. Enzymatic synthesis of novel purine nucleosides bearing a chiral benzoxazine fragment. Chem. Biol. Drug Design. 2019, 93, 605–616. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Musiyak, V.V.; Vozdvizhenskaya, O.A.; Galegov, G.A.; Andronova, V.L.; Gruzdev, D.A.; Chulakov, E.N.; Vigorov, A.Y.; Ezhikova, M.A.; Kodess, M.I.; et al. N-[omega-(Purin-6-yl)aminoalkanoyl] Derivatives of Chiral Heterocyclic Amines as Promising Anti-Herpesvirus Agents. Eur. J. Org. Chem. 2019, 2019, 4811–4821. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Levit, G.L.; Musiyak, V.V.; Gruzdev, D.A.; Charushin, V.N. Fragment-based approach to novel bioactive purine derivatives. Pure Appl. Chem. 2020, 92, 1277–1295. [Google Scholar] [CrossRef]
- Vozdvizhenskaya, O.A.; Andronova, V.L.; Galegov, G.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Synthesis and antiherpetic activity of novel purine conjugates with 7,8-difluoro-3-methyl-3,4-dihydro-2H-1,4-benzoxazine. Chem. Heterocycl. Compd. 2021, 57, 490–497. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Zarubaev, V.V.; Gruzdev, D.A.; Vozdvizhenskaya, O.A.; Vakarov, S.A.; Musiyak, V.V.; Chulakov, E.N.; Volobueva, A.S.; Sinegubova, E.O.; Ezhikova, M.A.; et al. Novel purine–N-heterocycle conjugates: Synthesis and anti-influenza activity. Chem. Heterocycl. Compd. 2021, 57, 498–504. [Google Scholar] [CrossRef]
- Krasnov, V.P.; Musiyak, V.V.; Levit, G.L.; Gruzdev, D.A.; Andronova, V.L.; Galegov, G.A.; Orshanskaya, I.R.; Sinegubova, E.O.; Zarubaev, V.V.; Charushin, V.N. Synthesis of Pyrimidine Conjugates with 4-(6-Aminohexanoyl)-7,8-difluoro-3,4-dihydro-3-methyl-2H-[1,4]benzoxazine and Evaluation of Their Antiviral Activity. Molecules 2022, 27, 4236. [Google Scholar] [CrossRef]
- Ward, D.N.; Wade, J.; Walborg, E.F., Jr.; Osdene, T.S. The Synthesis of N-(6-Purinyl)amino Acids. Amino Acids with a Single Reactive Amino Group. J. Org. Chem. 1961, 26, 5000–5005. [Google Scholar] [CrossRef]
- Frydrych, J.; Poštová-Slavětínská, L.; Dračínský, M.; Janeba, Z. Efficient Synthesis of α-Branched Purine-Based Acyclic Nucleosides: Scopes and Limitations of the Method. Molecules 2020, 25, 4307. [Google Scholar] [CrossRef]
- Slepukhin, P.A.; Gruzdev, D.A.; Chulakov, E.N.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Structures of the racemate and (S)-enantiomer of 7,8-difluoro-3-methyl-2,3-dihydro-4H-[1,4]benzoxazine. Russ. Chem. Bull. 2011, 60, 955–960. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; DeJesus, R.E.; Contreras, L.; Rodriguez-Palomares, I.A.; Villanueva, P.J.; Balderrama, K.S.; Monterroza, L.; Larragoity, M.; Varela-Ramirez, A.; Aguilera, R.J. A new pyridazinone exhibits potent cytotoxicity on human cancer cells via apoptosis and poly-ubiquitinated protein accumulation. Cell Biol. Toxicol. 2019, 35, 503–519. [Google Scholar] [CrossRef]
- Mfotie Njoya, E.; Maza, H.L.D.; Mkounga, P.; Koert, U.; Nkengfack, A.E.; McGaw, L.J. Selective cytotoxic activity of isolated compounds from Globimetula dinklagei and Phragmanthera capitata (Loranthaceae). Z. Naturforsch. C 2020, 75, 135–144. [Google Scholar] [CrossRef]
- Rahmé, R. Assaying Cell Cycle Status Using Flow Cytometry. Methods Mol. Biol. 2021, 2267, 165–179. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Compound | |||||||
|---|---|---|---|---|---|---|---|---|
| 1c | 1d | 1e | 1f | |||||
| CC50 | SCI | CC50 | SCI | CC50 | SCI | CC50 | SCI | |
| WI-38 | 50 | - | 21 | - | 71 | - | 35 | - |
| CT-26 | 19 | 2.6 | 19 | 1.1 | 3.4 | 21 | 3.0 | 12 |
| 4T1 | 23 | 2.2 | 23 | 0.91 | 0.49 | 145 | 2.3 | 15 |
| MDA-MB-231 | 44 | 1.2 | 44 | 0.48 | 12 | 5.9 | 28 | 1.4 |
| COLO201 | 0.68 | 73 | 0.68 | 31 | 3.5 | 20 | 1.3 | 27 |
| HepG2 | 8.5 | 5.9 | 8.5 | 2.5 | 5.2 | 14 | 9.7 | 3.6 |
| A549 | 43 | 1.2 | 43 | 0.49 | 75 | 0.95 | 60 | 0.58 |
| SK-BR-3 | 75 | 0.67 | 75 | 0.28 | 85 | 0.83 | 99 | 0.35 |
| SNU-1 | 7.0 | 7.1 | 7.0 | 3.0 | 6.0 | 12 | 6.0 | 5.8 |
| Jurkat | 8.5 | 5.9 | 8.5 | 2.5 | 4.0 | 18 | 32 | 1.1 |
| Compound | CT-26 | 4T1 | MDA-MB-231 | COLO201 | HepG2 | A549 | SK-BR-3 | SNU-1 | Jurkat | WI-38 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1c | 2 ± 1 | 2 ± 2 | 5 ± 5 | 2 ± 1 | 2 ± 1 | 7 ± 1 | 8 ± 0 | 6 ± 2 | 8 ± 2 | 21 ± 3 |
| 1d | 2 ± 1 | 2 ± 1 | 4 ± 2 | 1 ± 1 | 2 ± 1 | 6 ± 1 | 8 ± 2 | 5 ± 1 | 10 ± 4 | 21 ± 7 |
| 1e | 3 ± 2 | 2 ± 2 | 7 ± 5 | 2 ± 1 | 2 ± 1 | 6 ± 0 | 9 ± 2 | 5 ± 1 | 10 ± 4 | 22 ± 7 |
| Compound | Cell Lines | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| WI-38 | A549 | SK-BR-3 | SNU-1 | Jurkat | |||||
| CC50 | CC50 | SCI | CC50 | SCI | CC50 | SCI | CC50 | SCI | |
| 1a | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 |
| 1b | 9.8 × 10–5 | 5.3 × 10–5 | 1.8 | >1.0 × 10–4 | 0.98 | 2.0 × 10–5 | 4.9 | 6.7 × 10–5 | 1.5 |
| (S)-1b | 8.1 × 10–5 | 5.8 × 10–4 | 0.14 | >1.0 × 10–4 | 0.81 | 2.6 × 10–5 | 3.1 | 3.7 × 10–5 | 2.2 |
| 1d | 2.1 × 10–5 | 2.1 × 10–5 | 1.0 | 2.8 × 10–5 | 0.75 | 2.0 × 10–6 | 10 | 1.0 × 10–5 | 2.1 |
| 2a | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | 5.9 × 10–5 | 1.7 |
| 2b | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | 4.9 × 10–5 | 2.0 |
| 2c | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 |
| 6 | 3.0 × 10–5 | 1.5 × 10–5 | 2.0 | 3.7 × 10–4 | 0.08 | 5.6 × 10–6 | 5.4 | 1.0 × 10–5 | 3.0 |
| 7 | 1.4 × 10–5 | 9.4 × 10–6 | 1.5 | 4.2 × 10–5 | 0.33 | 3.7 × 10–6 | 3.8 | 7.3 × 10–6 | 1.9 |
| (S)-9 | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 |
| 9 | >1.0 × 10–4 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 | >1.0 × 10–4 | 1.0 |
| Dox | 2.3 × 10–7 | 2.2 × 10–7 | 1.0 | 1.5 × 10–6 | 0.15 | 1.4 × 10–7 | 1.6 | 1.2 × 10–7 | 1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasnov, V.P.; Vozdvizhenskaya, O.A.; Baryshnikova, M.A.; Pershina, A.G.; Musiyak, V.V.; Matveeva, T.V.; Nevskaya, K.V.; Brikunova, O.Y.; Gruzdev, D.A.; Levit, G.L. Synthesis and Cytotoxic Activity of the Derivatives of N-(Purin-6-yl)aminopolymethylene Carboxylic Acids and Related Compounds. Molecules 2023, 28, 1853. https://doi.org/10.3390/molecules28041853
Krasnov VP, Vozdvizhenskaya OA, Baryshnikova MA, Pershina AG, Musiyak VV, Matveeva TV, Nevskaya KV, Brikunova OY, Gruzdev DA, Levit GL. Synthesis and Cytotoxic Activity of the Derivatives of N-(Purin-6-yl)aminopolymethylene Carboxylic Acids and Related Compounds. Molecules. 2023; 28(4):1853. https://doi.org/10.3390/molecules28041853
Chicago/Turabian StyleKrasnov, Victor P., Olga A. Vozdvizhenskaya, Maria A. Baryshnikova, Alexandra G. Pershina, Vera V. Musiyak, Tatyana V. Matveeva, Kseniya V. Nevskaya, Olga Y. Brikunova, Dmitry A. Gruzdev, and Galina L. Levit. 2023. "Synthesis and Cytotoxic Activity of the Derivatives of N-(Purin-6-yl)aminopolymethylene Carboxylic Acids and Related Compounds" Molecules 28, no. 4: 1853. https://doi.org/10.3390/molecules28041853