
Effect of Acetylation on the Nanofibril Formation of Chitosan from All-Atom De Novo Self-Assembly Simulations


Abstract
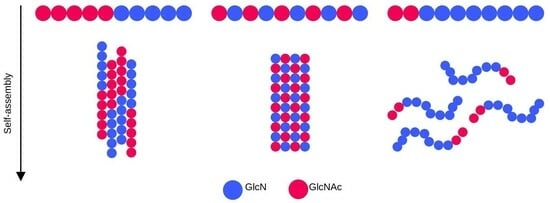
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the Molecular Mechanics Force Field and Summary of Simulations
2.2. Chitosan Solubility and Self-Assembly Is Dependent on the Degree of Acetylation
2.3. Pattern of Acetylation Influences Chitosan’s Self-Assembly Process
2.4. The Self-Assembled Nanofibrils Are Comprised of Nearly Exclusively Antiparallel Chains Regardless of the Acetylation Pattern
2.5. Intermolecular Hydrogen Bonding Pattern Is Similar for the Nanofibrils with Block and Alternating PAs
3. Concluding Discussion
4. Materials and Methods
4.1. Force Field Parameterization
4.2. Molecular Dynamics Simulation
4.3. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinaudo, M. Chitin and chitosan: Properties and applcations. Progress in Polymer Science. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Kumar, M.N.V.R.; Muzzarelli, R.A.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gomez, C.P.; Cecilia, J.A. Chitosan: A natural biopolymer with a wide and varied range of applications. Molecules 2020, 25, 3981. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, M.X.; Sauvageau, J.C.; Kumirska, J.; Thoming, J. Studies on acetylation patterns of different chitosan preparations. Carbohydr. Polym. 2009, 78, 678–684. [Google Scholar] [CrossRef]
- Wattjes, J.; Sreekumar, S.; Richter, C.; Cord-Landwehr, S.; Singh, R.; El Geuddari, N.; Moerschbacher, B.M. Patterns matter part 1: Chitosan polymers with non-random patterns of acetylation. React. Funct. Polym. 2020, 151, 104583. [Google Scholar] [CrossRef]
- Wattjes, J.; Niehues, A.; Cord-Landwehr, S.; Hoßbach, J.; David, L.; Delair, T.; Moerschbacher, B.M. Enzymatic production and enzymatic-mass spectrometric fingerprinting analysis of chitosan polymers with different nonrandom patterns of acetylation. J. Am. Chem. Soc. 2019, 141, 3137–3145. [Google Scholar] [CrossRef]
- Kaczmarek, M.B.; Struszczyk-Swita, K.; Li, X.; Szczęsna-Antczak, M.; Daroch, M. Enzymatic modifications of chitin, chitosan, and chitooligosaccharides. Front. Bioeng. Biotechnol. 2019, 7, 243. [Google Scholar] [CrossRef]
- Sreekumar, S.; Wattjes, J.; Niehues, A.; Mengoni, T.; Mendes, A.C.; Morris, E.R.; Goycoolea, F.M.; Moerschbacher, B.M. Biotechnologically produced chitosans with nonrandom acetylation patterns differ from conventional chitosans in properties and activities. Nat. Commun. 2022, 13, 7125. [Google Scholar] [CrossRef]
- Basa, S.; Nampally, M.; Honorato, T.; Das, S.N.; Podile, A.R.; El Gueddari, N.E.; Moerschbacher, B.M. The pattern of acetylation defines the priming activity of chitosan tetramers. J. Am. Chem. Soc. 2020, 142, 1975–1986. [Google Scholar] [CrossRef]
- Foster, L.J.R.; Ho, S.; Hook, J.; Basuki, M.; Marçal, H. Chitosan as a biomaterial: Influence of degree of deacetylation on its physiochemical, material and biological properties. PLoS ONE 2015, 10, e0135153. [Google Scholar] [CrossRef]
- Cao, W.; **g, D.; Li, J.; Gong, Y.; Zhao, N.; Zhang, Z. Effects of the degree of deacetylation on the physicochemical properties and schwann cell affinity of chitosan films. J. Biomater. Appl. 2005, 20, 157–177. [Google Scholar]
- Chatelet, C.; Damour, O.; Domard, A. Influence of the degree of acetylation on some biological properties of chitosan films. Biomaterials 2001, 22, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Franca, E.F.; Freitas, L.C.G.; Lins, R.D. Chitosan molecular structure as a function of N-acetylation. Biopolymers 2011, 95, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Tsereteli, L.; Grafmüller, A. An accurate coarse-grained model for chitosan polysaccharides in aqueous solution. PLoS ONE 2017, 12, e0180938. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Hatcher, E.; Venable, R.M.; Pastor, R.W.; MacKerell, A.D. Charmm additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W.; MacKerell, A.D. Charmm additive all-atom force field for carbohydrate derivatives and its Utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef]
- Morrow, B.H.; Payne, G.F.; Shen, J. pH-responsive self-assembly of polysaccharide through a rugged energy landscape. J. Am. Chem. Soc. 2015, 137, 13024–13030. [Google Scholar] [CrossRef]
- Tsai, C.C.; Morrow, B.H.; Chen, W.; Payne, G.F.; Shen, J. Toward understanding the environmental control of hydrogel film properties: How salt modulates the flexibility of chitosan chains. Macromolecules 2017, 50, 5946–5952. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the Charmm general force field (cgenff) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Guvench, O.; Greene, S.N.; Kamath, G.; Brady, J.W.; Venable, R.M.; Pastor, R.W.; Mackerell, A.D. Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 2008, 29, 2543–2564. [Google Scholar] [CrossRef]
- Schatz, C.; Viton, C.; Delair, T.; Pichot, C.; Domard, A. Typical physicochemical behaviors of chitosan in aqueous solution. Biomacromolecules 2003, 4, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Romany, A.; Payne, G.F.; Shen, J. Mechanism of the temperature-dependent self-assembly and polymorphism of chitin. Chem. Mater. 2023, 35, 6472–6481. [Google Scholar] [CrossRef]
- Lamarque, G.; Lucas, J.M.; Viton, C.; Domard, A. Physicochemical behavior of homogeneous series of acetylated chitosans in aqueous solution: Role of various structural parameters. Biomacromolecules 2005, 6, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.A.; Shen, J.K. Probing the strand orientation and registry alignment in the propagation of amyloid fibrils. Biochemistry 2010, 49, 5290–5298. [Google Scholar] [CrossRef]
- Naito, P.K.; Ogawa, Y.; Sawada, D.; Nishiyama, Y.; Iwata, T.; Wada, M. X-ray crystal structure of anhydrous chitosan at atomic resolution. Biopolymers 2016, 105, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, K.; Noguchi, K.; Miyazawa, T.; Yui, T.; Ogawa, K. Molecular and crystal structure of hydrated chitosan. Macromolecules 1997, 30, 5849–5855. [Google Scholar] [CrossRef]
- Lertworasirikul, A.; Yokoyama, S.; Noguchi, K.; Ogawa, K.; Okuyama, K. Molecular and crystal structures of chitosan/HI type I salt determined by x-ray fiber diffraction. Carbohydr. Res. 2004, 339, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Yui, T.; Imada, K.; Okuyama, K.; Obata, Y.; Suzuki, K.; Ogawa, K. Molecular and crystal structure of the anhydrous form of chitosan. Macromolecules 1994, 27, 7601–7605. [Google Scholar] [CrossRef]
- Mazeau, K.; Winter, W.T.; Chanzy, H. Molecular and crystal structure of a high-temperature polymorph of chitosan from electron diffraction data. Macromolecules 1994, 27, 7606–7612. [Google Scholar] [CrossRef]
- Okuyama, K.; Noguchi, K.; Hanafusa, Y.; Osawa, K.; Ogawa, K. Structural study of anhydrous tendon chitosan obtained via chitosan/acetic acid complex. Int. J. Biol. Macromol. 1999, 26, 285–293. [Google Scholar] [CrossRef]
- Jang, M.K.; Kong, B.G.; Jeong, Y.I.; Lee, C.H.; Nah, J.W. Physicochemical characterization of α-chitin, β-chitin, and γ-chitin separated from natural resources. J. Polym. Sci. A Polym. Chem. 2004, 42, 3423–3432. [Google Scholar] [CrossRef]
- Fan, Y.; Saito, T.; Isogai, A. Preparation of chitin nanofibers from squid pen β-chitin by simple mechanical treatment under acid conditions. Biomacromolecules 2008, 9, 1919–1923. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Mujtaba, M.; Ehrlich, H.; Salaberria, A.M.; Baran, T.; Amemiya, C.T.; Galli, R.; Akyuz, L.; Sargin, I.; Labidi, J. On chemistry of γ-chitin. Carbohydr. Polym. 2017, 176, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Kumar, A.; Yoluk, O.; MacKerell, A.D., Jr. FFParam: Standalone package for CHARMM additive and DRUDE polarizable force field parametrization of small molecules. J. Comput. Chem. 2020, 41, 958–970. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2020. Available online: https://ambermd.org/Manuals.php (accessed on 14 January 2024).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Martinez, L.; Andrade, R.; Birgin, E.; Martinez, J. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Brooks, B.; Brooks, C.; MacKerell, A.; Nilsson, L.; Petrella, R.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Grønbech-Jensen, N.; Farago, O. A simple and effective verlet-type algorithm for simulating langevin dynamics. Mol. Phys. 2013, 111, 983–991. [Google Scholar] [CrossRef]
- Chow, K.H.; Ferguson, D.M. Isothermal-isobaric molecular dynamics simulations with monte carlo volume sampling. Comput. Phys. Commun. 1995, 91, 283–289. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DA | PA | DP | Chains | Simulation Time |
|---|---|---|---|---|
| 20% | Block | 10 | 24 | 3 × 2 s |
| 50% | Block | 10 | 24 | 3 × 3 s |
| 50% | Alternating | 10 | 24 | 2 × 4 s, 4.5 s |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romany, A.; Payne, G.F.; Shen, J. Effect of Acetylation on the Nanofibril Formation of Chitosan from All-Atom De Novo Self-Assembly Simulations. Molecules 2024, 29, 561. https://doi.org/10.3390/molecules29030561
Romany A, Payne GF, Shen J. Effect of Acetylation on the Nanofibril Formation of Chitosan from All-Atom De Novo Self-Assembly Simulations. Molecules. 2024; 29(3):561. https://doi.org/10.3390/molecules29030561
Chicago/Turabian StyleRomany, Aarion, Gregory F. Payne, and Jana Shen. 2024. "Effect of Acetylation on the Nanofibril Formation of Chitosan from All-Atom De Novo Self-Assembly Simulations" Molecules 29, no. 3: 561. https://doi.org/10.3390/molecules29030561