1. Introduction
In the long domestication history of barley, many interesting traits such as non-brittle rachis, six-rowed spike and naked caryopsis appeared subsequently [
1]. Among these domestication traits, the covered (hulled) or naked (hulless) caryopsis character of barley is an important agronomic trait for its linking to use purposes directly. Most cultivars have covered caryopses as animal feed and for brewing in which the hull (outer lemma and inner palea) and the pericarp epidermis are adhered firmly at maturity. However, a few cultivars are of a free-threshing varieties as human food in which the hull is departed from caryopsis at maturity easily [
2]. Tibetan naked barley, is an important conventional zanba food crop which is widely cultivated and has abundant germplasm resource in the Qinghai-Tibet Plateau, China [
3,
4].
Originally Harlan and Hulton reported that a sticky adhesive substance appears 10 days after flowering on the caryopsis surface in hulled barley [
5]. Transmission electron microscopy showed that this substance is secreted from the pericarp epidermis two days after flowering and its thickness increases during grain development [
6]. Taketa and his colleagues reported that this substance is cuticular lipid, and the presence or absence of the lipid layer on the pericarp epidermis is critical for distinguishing covered and naked barley [
7,
8,
9]. A single recessive gene,
nud, located on chromosome 7HL [
10], was found to control the naked caryopsis character, suggesting that easy separation of the hull results from a
nud mutation that damaged gene function. Through positional cloning, the
Nud (allele for covered caryopsis) gene is found to encode an ethylene response factor (ERF) family transcription factor and shows high similarity to the Arabidopsis WIN1/SHN1 transcription factor gene, whose deduced function is control of a lipid biosynthesis pathway [
9]. However, the molecular mechanism underlying this hulled/naked character remains largely unknown.
Cuticular waxes and cutin form the cuticle, a hydrophobic layer covering the aerial surfaces of land plants and acting as a protective barrier against environmental stresses. So far, about 55 genes involved in the formation of the cuticle have been isolated, which are divided into three categories: regulatory genes, synthesis genes, and transporter genes [
11,
12,
13,
14]. In barley, >1580
eceriferum (
cer) mutants with different degrees of reduced wax in different parts, are classified into 79 loci [
15,
16]. Three barley
cer mutants (
cer-yl,
cer-ym and
cer-zv) show weak hull-caryopsis adhesion [
17], suggesting a link between epidermal cuticle lipids and hull-caryopsis adhesion [
9].
In order to further understand the molecular mechanism of covered/naked caryopsis trait, comparative transcriptome profile of develo** caryopses of Tibetan Hulless barley landrace “Dulihuang” and covered barley cultivar “Morex” were analyzed by RNA-seq technique. We found that the NUD transcription factor might down regulate cutcle biosynthesis genes in covered barley, and that deletion or low expression level of the Nud gene may result in the normal expression of cuticle related genes in naked barley.
3. Discussion
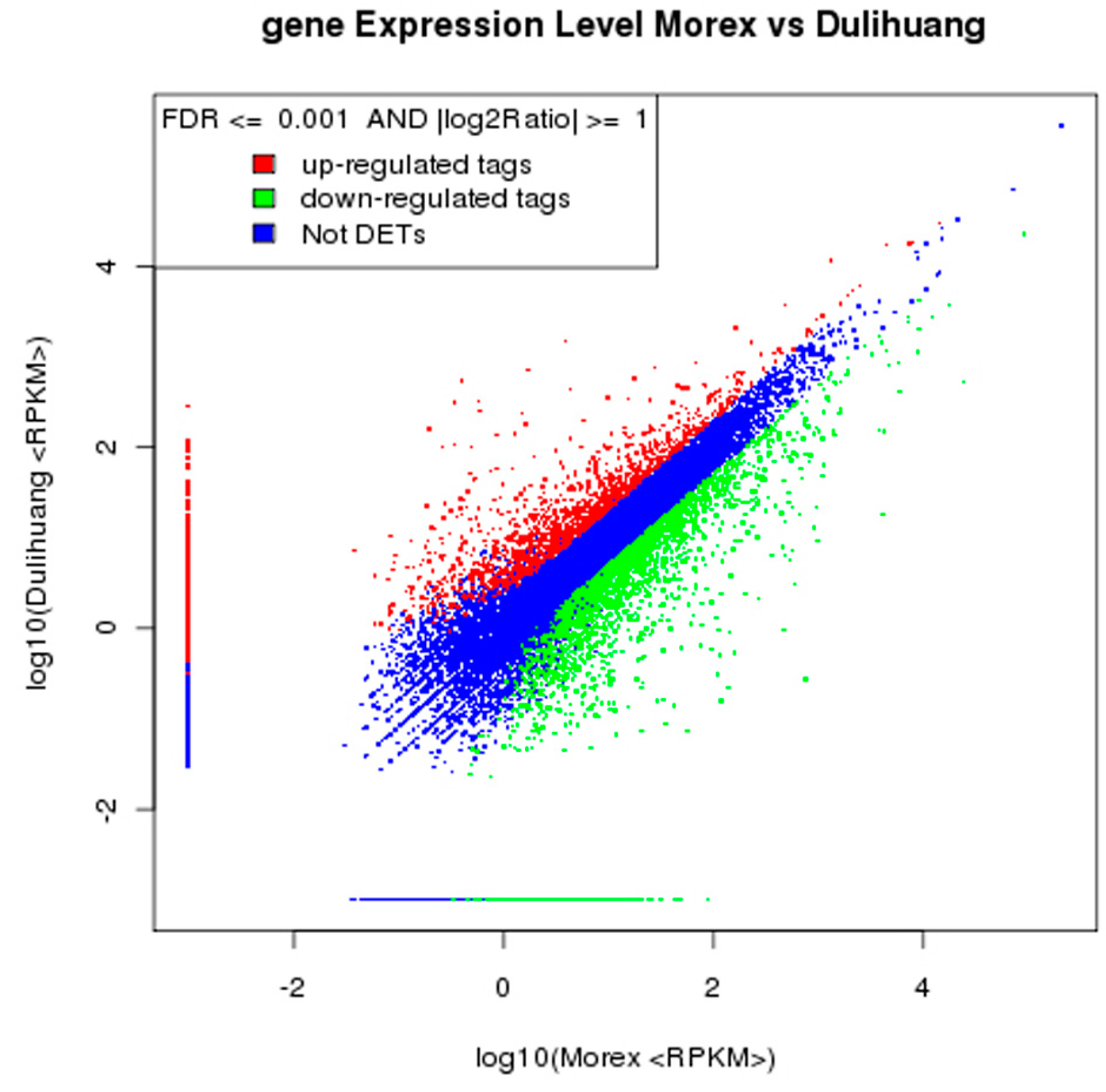
We carried out a transcriptome analysis of caryopses of hulled and hulless barley, Morex and Duliuang, respectively, to reveal genes that may be involved in hull-caryopsis adhesion. A total of 4031 DEGs were identified, most of which were classified into three groups, metabolic process, cellular process, and response to stimulus, in biological process category of gene ontology (GO) functional classification. The same three top groups are also found in DEGs in caryopses of two Tibetan hulless barley varieties [
19], indicating that the majority of DEGs in the present study were not related to covered/naked caryopsis trait. Most of the DEGs may reflect many other caryopsis traits varied between Morex and Dulihuang. A cementing layer causes hull-caryopsis adhesion in covered caryopses [
9]. This cementing layer is a thickened and loose cuticle of pericarp [
6,
20]. Thus, we focused on DEGs associated with cuticle development.
Expression level of
Nud gene was much more pronounced in Morex than in Dulihuang. This result confirms that the
Nud gene is responsible for hull-caryopsis adhesion in hulled barley [
9]. NUD protein is a transcription factor related to regulation of lipid biosynthesis pathway. It may regulate expression of genes involved in the development of pericarp cuticle in hulled barley, but not in hulless barley, because the
Nud gene is deleted in all 100 hulless barley cultivars tested [
9] and the origin of hulless barley is monophyletic [
7,
9]. To our surprise, the
Nud gene was found to be expressed in hulless barley cultivar Dulihuang. We investigated caryopsis RNA-seq results (NCBI Sequence Read Archive, Accession numbers: SRR1032035, SRR1032036, SRX375649 and SRX378862) of two Tibetan hulless barley cultivars [
19], and found that the
Nud gene was also expressed in one of them. The genomic sequence of the
Nud gene in Dulihuang and the other hulless barley will be analyzed to confirm its existence in hulless barley. There might be mutations in the two
Nud sequences or posttranslational regulation of the two NUD protein, which lead to the naked phenotype.
Table 2.
Differentially expressed cuticle related genes.
Table 2.
Differentially expressed cuticle related genes.
| Gene Name | Gene ID | Plant | Function | Mutant Phenotype | Barley Gene | Log2 Ratio (Morex/Dulihuang) | FDR |
|---|
| | Cuticle Permeability | Organ Fusion |
|---|
| Nud | GU108424 | Barley | Regulation | No | No | MLOC_59305.1 | +5.13 | 5.78 × 10−18 |
| SHN1 | At1g15360 | Arabidopsis | Regulation | Permeable | Organ fusion | MLOC_12747.2 | −3.12 | 2.48 × 10−10 |
| ATT1 | AT4G00360 | Arabidopsis | Cutin biosynthesis | Permeable | No | MLOC_71597.1 | −2.53 | 2.84 × 10−273 |
| GPAT6 | AT2G38110 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_56115.1 | −1.93 | 8.71 × 10−51 |
| GPAT8 | AT4G00400 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_19148.1 | −4.38 | 1.36 × 10−5 |
| LACS2 | AT1G49430 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_66827.1 | −2.42 | 1.15 × 10−39 |
| LCR | AT2G45970 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_75020.2 | −1.37 | 2.27 × 10−7 |
| HTH | AT1G72970 | Arabidopsis | Cutin biosynthesis | Permeable | Organ fusion | MLOC_9891.3 | −1.78 | 1.89 × 10−6 |
| FDH1 | AT2G26250 | Arabidopsis | Wax biosynthesis | Permeable | Organ fusion | MLOC_65700.1 | −1.96 | 1.47 × 10−280 |
| WAX2 | AY131334 | Arabidopsis | Unknown | Permeable | Organ fusion | MLOC_51499.1 | −1.05 | 2.16 × 10−6 |
| CER9 | At4g34100 | Arabidopsis | Unknown | No | No | MLOC_36840.2 | +1.10 | 5.29 × 10−46 |
| ACC1 | AT1G36160 | Arabidopsis | Wax biosynthesis | Permeable | Organ fusion | MLOC_37244.1 | −1.63 | 5.61 × 10−47 |
| EIBI1 | AB534898 | Barley | Secretion | Permeable | - | MLOC_62487.1 | −1.63 | 5.43 × 10−86 |
| WBC11 | At1g17840 | Arabidopsis | Secretion | Permeable | Organ fusion | AK355515 | −1.10 | 4.35 × 10−12 |
| MYB96 | At5G62470 | Arabidopsis | Regulation | No | No | MLOC_34885.1 | −1.72 | 5.80 × 10−9 |
| WSD1 | AT5G37300 | Arabidopsis | Wax biosynthesis | No | No | MLOC_74286.1 | −1.86 | 7.78 × 10−15 |
| CUT1 | AT1G68530 | Arabidopsis | Wax biosynthesis | No | No | MLOC_51583.2 | −2.36 | 4.61 × 10−34 |
| FATB | AT1G08510 | Arabidopsis | Fatty acids biosynthesis | No | No | MLOC_76304.1 | −1.21 | 0.00015 |
NUD might regulate one transcription factor gene (
SHN1), six cutin biosynthesis genes (
ATT1,
LCR,
GPAT6,
GPAT8,
LACS2, and
HTH), four cutin related genes (
FDH1,
WAX2,
CER9, and
ACC1), and two cutin transporter genes (
Eibi1 and
WBC11). SHN1 primarily controls cutin biosynthesis by regulating cutin related genes, and indirectly affect wax accumulation [
21]. ATT1 and LCR are most likely involved in the cutin monomer biosynthesis as cytochrome P450s of the CYP86A type [
22,
23]. GPAT6 and GPAT8 are sn-2 acyl transferase with glycerol-3-phosphate as the acyl acceptor [
24]. LACS2 is a long chain acyl-CoA synthetase required to activate free fatty acids to the acyl-CoAs [
25]. HTH is an oxidoreductase that is thought to be involved in the biosynthesis of dicarboxylic acids [
26]. Although FDH has been identified as a β-ketoacyl-CoA synthase based on sequence similarity [
27], its mutant
fdh shows an increase in cutin constituents [
28]. The biochemical role of FDH in cutin formation has not yet been elucidated [
29]. WAX2 is a protein of unknown function required for cuticular wax biosynthesis; it also may be necessary for cutin synthesis [
30]. CER9 is an E3 ubiquitin ligase; loss of its function leads to coordinated alterations in cutin and wax biosynthesis and improved drought resistance of the cuticle [
31]. ACC1, an acetyl coenzyme A carboxylase, plays a primary role in the biosynthesis of very-long-chain fatty acids associated with cuticular waxes; its mutant allele
acc1/
gsd1 has less amount of wax but more cutin deposition on inflorescence stems [
32]. Eibi1 is a full ABCG transporter identified in barley and WBC11 is a half ABCG transporter identified in Arabidopsis, both for cutin deposition [
33,
34]. Mutations (
shn1,
gpat6,
lacs2,
lcr,
hth,
fdh1,
wax2,
acc1 and
wbc11) in most of the above-mentioned genes, lead to two common phenotypes, enhanced cuticle permeability and organ fusion. NUD may down-regulates the expression of cutin related genes (
SHN1,
GPAT6,
GPAT8,
LACS2,
LCR,
HTH,
FDH1,
WAX2,
WBC11, and
ATT1) to make a permeable cuticle proper, which caused to the hull-caryopsis fusion in hulled barley.
CER9 is the only upregulated cutin linked gene in covered caryopses; it is a unique gene whose mutation improves hydrophobic barriers to water diffusion through cuticle membrane [
31]. An upregulation of
CER9 gene expression may cause a more permeable cuticle, which has advantages for hull-caryopsis fusion in hulled barley. Taken together, these data suggest that NUD regulation of cutin biosynthesis pathway may be involved in covered caryopsis development.
The pericarp cuticle of covered caryopsis (figure 37 in [
6]) is reminiscent of the cuticle proper of organ fusion mutants, such as
wax2 and
wbc11, which is thicker with a loose and less osmiophilic structure (figure 1 in [
30]; figure 7e in [
34]), while the pericarp cuticle of naked caryopsis (figure 9 in [
6]) is comparable to wild-type cuticle (figure 7d in [
34]). Insoluble cutin is the major structural material of plant cuticle [
35]. The thick and loose cutin is able to explain the increased permeability of pericarp cuticle in covered caryopses rather than in the naked one [
9]. High permeable cuticle of pericarp may cause fusion of the hull and caryopsis. We may conclude that hulled barley displays hull-caryopsis fusion instead of adhesion.
A naked caryopsis, without the hull protection, has to build up a functional cuticle to seal itself to prevent water loss. A transcription factor MYB96 was upregulated in hulless Dulihuang. MYB96 improves plant drought resistance by promoting cuticular wax accumulation under drought stress; it binds directly to the promoters of wax biosynthetic genes and subsequently activating their transcription. The upregulated wax biosynthetic genes by MYB96 include
WSD1,
FATB, and
CUT1 [
36]. These three genes were also upregulated in Dulihuang. We therefore propose that the MYB96 regulation of wax biosynthesis pathway may be involved in naked caryopsis development.
4. Experimental Section
4.1. Plant Material
For transcriptomic analyses, barley varieties Morex and Tibetan Hulless barley Landrace Dulihuang were used. Two varieties were planted in a field of Academy of Agriculture and Forestry, Qinghai University. At grain filling stage, three replicates of three-week-old caryopses (including hulls at three weeks after anthesis) from both varieties were collected for RNA extraction.
4.2. RNA Extraction, cDNA Library Preparation, and Sequencing
Total RNA was prepared using Trizol according to the manufacturer’s protocol (Invitrogen, Burlington, ON, Canada) with some modification. The concentration of each RNA sample was determined by NanoDrop 2000TM micro-volume spectrophotometer (Thermo Scientific, Waltham, MA, USA) and gel electrophoresis. The RNA quality was checked on a Bioanalyzer 2100 (Aligent, Santa Clara, CA, USA); RNA Integrity Number (RIN) values were greater than 8.5 for all samples.
Sequencing libraries were prepared according to the manufacturer’s instructions (Illumina, San Diego, CA, USA).
Primary sequencing data produced by Illumina HiSeqTM 2000 (Illumina, San Diego, CA, USA), called raw reads, is subjected to quality control (QC) to determine if a resequencing step is needed. The raw reads were cleaned by removing adaptor sequences, empty reads and low quality sequences.
During the quality control (QC) steps, the 2100 Bioanylzer (Agilent Technologies, Santa Clara, CA, USA) and ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) were used in quantification and qualification of the sample library. Finally, the library is sequenced using Illumina HiSeqTM 2000.
4.3. Functional Annotation: Evaluation of Genes from RNA-Seq
After QC, raw reads were filtered into clean reads to be aligned to the barley reference sequences with SOAPaligner/SOAP2 [
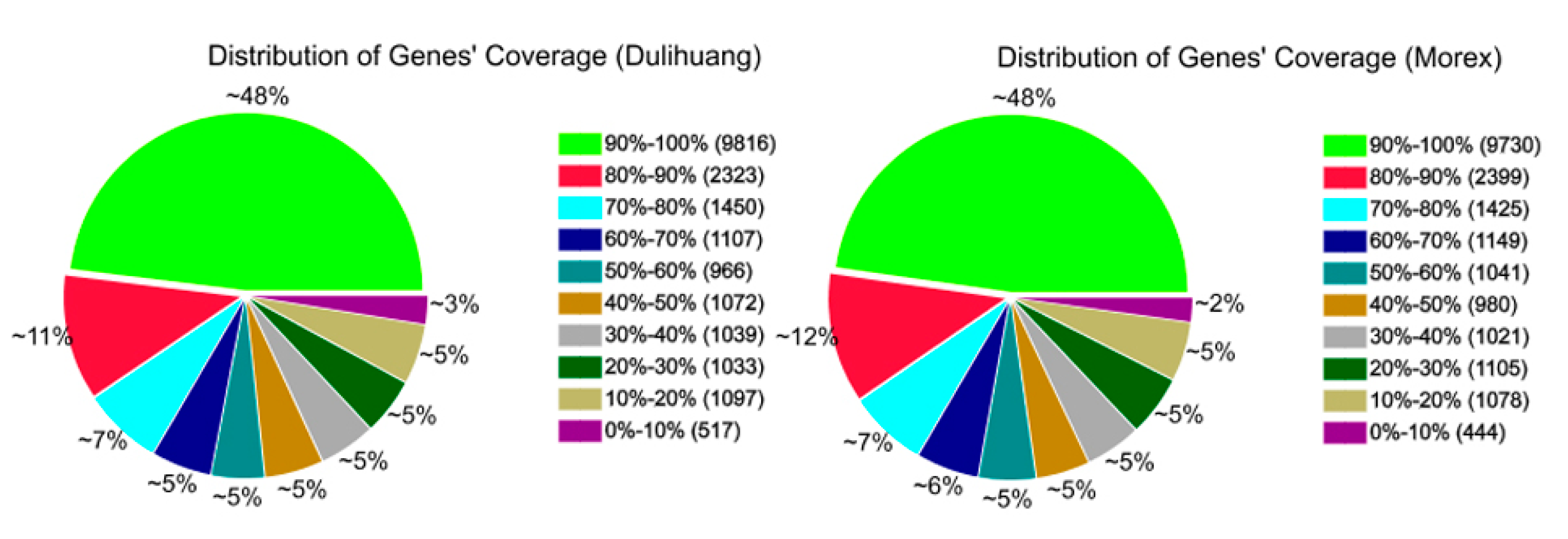
37]. The alignment data was utilized to calculate distribution of reads on reference genes and perform gene coverage analysis. After results have passed QC, downstream analysis including gene expression and gene annotation were performed. Results of gene expression include gene expression levels and differential expression analysis. Further, we perform Gene Ontology (GO) enrichment analysis.
Raw digital gene expression counts were normalized by a variation of the RPKM (Reads per kb per million reads) method, which could eliminate the influence of different gene length and sequence discrepancy on the calculation of gene expression. Therefore, the calculated gene expression could be directly used for comparing the difference of gene expression among samples. For all RPKM values of each gene, the cutoff value for determining gene transcriptional activity was determined based on a 95% confidence threshold. The Blast2GO program was used to obtain GO annotations (version 2.3.5) for the all genes [
38]. Then, the software WEGO was used to perform GO functional classification of all genes to view the distribution of gene functions of the species at the macro level with the default parameters with a robust FDR correction to get an adjusted
p value [
39].
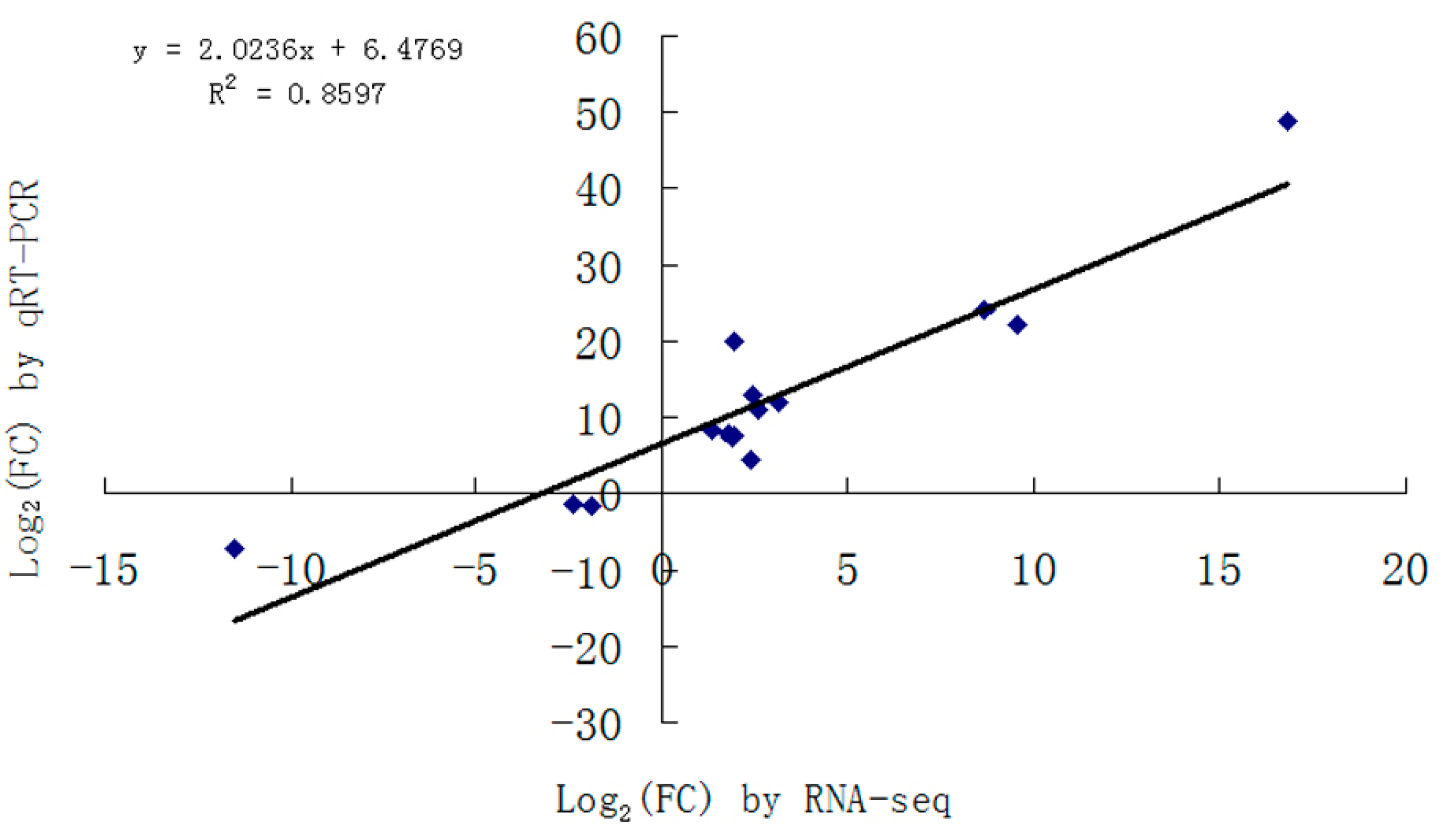
4.4. Validation of RNA-Seq by qRT-PCR
Total RNA from two samples (Morex and Dulihuang) was treated with DNase, and first-strand cDNA was generated using an AMV First Strand cDNA Synthesis Kit (Sangon, Shanghai, China). SYBR-based qRT-PCR reactions (SYBR Green I, ABI, Foster City, CA, USA) were performed on a LightCycler 480 system (Roche, Basel, Switzerland) using the following reaction conditions: 95 °C for 3 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 40 s. All qRT-PCR reactions were performed in triplicate, and the results were analyzed with the system’s relative quantification software (ver.1.5) based on the 2
−∆∆Ct method (Roche). The primers were listed in
Table S3. The detection threshold cycle for each reaction was normalized against the expression level of the HvActin gene with primer sequences 5′-AAGTACAGTGTCTGGATTGGAGGG-3′ and 5′-TCGCAACTTAGAAGCACTTCCG-3′.



{kind=link}
{kind=link}
{kind=link}


