1. Introduction
Gene expression characterization at the steady-state mRNA level is an important step in many plant biological processes. Northern blotting [
1,
2,
3,
4], is not an optimal method for gene expression quantification anymore, because it requires a large amount of RNA and is limited to detecting high-abundance transcripts. Therefore, reverse transcription-quantitative polymerase chain reaction (RT-qPCR), which can detect low-abundance mRNAs and exhibits high specificity, sensitivity, and stability [
5,
6,
7], has become one of the most commonly used techniques [
8]. However, RT-qPCR has certain disadvantages; for example, reference genes are important to correct the experimental error that occurs during RNA extraction, and cDNA preparation, but the normalized results are highly dependent on the expression stability of the reference genes selected [
9]. The use of inappropriate reference genes can result in biased target gene expression profiles; thus, using suitable reference genes for data normalization is a prerequisite for any RT-qPCR study [
10]. The expression of an ideal reference gene should not vary between different tissues or cells under investigation, nor in response to any experimental treatment [
11]. However, extensive transcriptomic data mining and gene expression analysis studies have reported that the reliability of these endogenous controls can be influenced by various conditions [
12,
13,
14,
15,
16,
17]. Therefore, to obtain accurate and reliable target gene expression profiles, it is important to identify the best reference genes for use in each experimental set-up and verify these in each individual experiment before performing gene expression normalization. In common plant species, including
Arabidopsis, rice, soybean, and petunia, genes associated with basic cellular processes such as those related to ubiquitin degradation, polyubiquitin, ubiquitin-conjugating enzymes, and ubiquitin ligases (collectively known as
UBQ), as well elongation factors (EFs) [
18,
19,
20,
21], have been identified and used for RT-qPCR [
6,
12,
13,
19]. Nevertheless, few reference genes have been identified in forest trees, which dominate much of the terrestrial landscape on earth. Members of the genus
Populus are used as a model forest species for diverse studies because of their amenability to experimental and genetic manipulation [
22,
23,
24,
25]. Studies in
Populus focus mainly on improving their ability to respond to environmental stress while maintaining their high-speed growth capacity because there is strong demand for their cultivation in highly saline and arid soils in many parts of the world [
26,
27].
Populus euphratica Oliv., which is native to desert regions ranging from western China to North Africa, is the only arboreal species established in the world’s largest shifting-sand desert, the Taklimakan Desert in China [
28]. The most significant characteristic of
P. euphratica is its extraordinary adaptation to salt stress [
28,
29,
30]. This species can survive concentrations of NaCl in nutrient solution up to 450 mM while maintaining higher growth and photosynthetic rates than other poplar species [
31,
32,
33]. Previous studies of
P. euphratica have mainly focused on analyses of salt-responsive genes [
26,
30,
34,
35], miRNAs [
36,
37], or salt-related transcriptome sequencing [
26,
38,
39]. In these studies, the genes up-regulated or down-regulated by salt stress are important and should be identified using validated reference genes by RT-qPCR.
Here, we evaluated the expression stability in
P. euphratica leaves, stems, and roots under salt stress of ten previously used reference genes [
40,
41], which are 60S ribosomal protein L35 (
60S),
Actin, elongation factor-1 α (
EF1α), eukaryotic initiation factor 5A (
eIF-5A), glyceraldehyde-3-phosphate dehydrogenase (
GAPDH), glucosidase II α-subunit (
GIIα), histone superfamily protein H3 (
HIS), ribosomal L27e protein family (
RP), tubulin β chain (
TUB) and ubiquitin family 6 (
UBQ). Five different methods were used for identification of suitable reference genes and the expression normalized by the reference genes selected by these methods was investigated for nine
P. euphratica genes; namely,
PeCOBL4 (COBRA-like extracellular glycosyl-phosphatidyl inositol-anchored protein family 4),
PeFLA12-1,
PeFLA12-2,
PeFLA12-3 and
PeFLA12-4 (FASCICLIN-like arabinogalactan-protein 12),
PeHKT1 (high-affinity K
+ transporter 1),
PeKUP3 (K
+ uptake transporter 3),
PeNhaD1 (NhaD-type Na
+/H
+ antiporter 1), and
PeNHX2 (Na
+/H
+ exchanger 2). The analyses show that the different reference genes identified by different methods had a variable impact on gene of interest data, and that a reliable set of reference genes should be selected based on the highest possible accuracy of RT-qPCR results. This approach can be applied to future studies of gene expression profiles in
P. euphratica.
3. Discussion
Although powerful techniques, including microarrays and high-throughput measurements, have been developed to detect gene expression levels, RT-qPCR is commonly used in many laboratories. Reliable RT-qPCR data depends on suitable reference genes, which must have highly stable expression under different experimental conditions. The validation of suitable reference genes is crucial, and has been performed previously in plants [
6,
12,
13,
18,
19,
20]. However, in woody plants the identification of reference genes is limited to maritime pine [
47], conifers [
48],
Eucalyptus [
49],
Jatropha curcas [
50] and two species of
Populus, namely,
P. trichocarpa ×
Populus deltoids [
51] and
Populus ×
euramericana [
40]. To facilitate the genetic improvement of woody plants for growth in saline soils, we performed reference gene evaluation in the salt tolerant tree
P. euphratica and found that none of the reference genes showed perfectly stable expression (
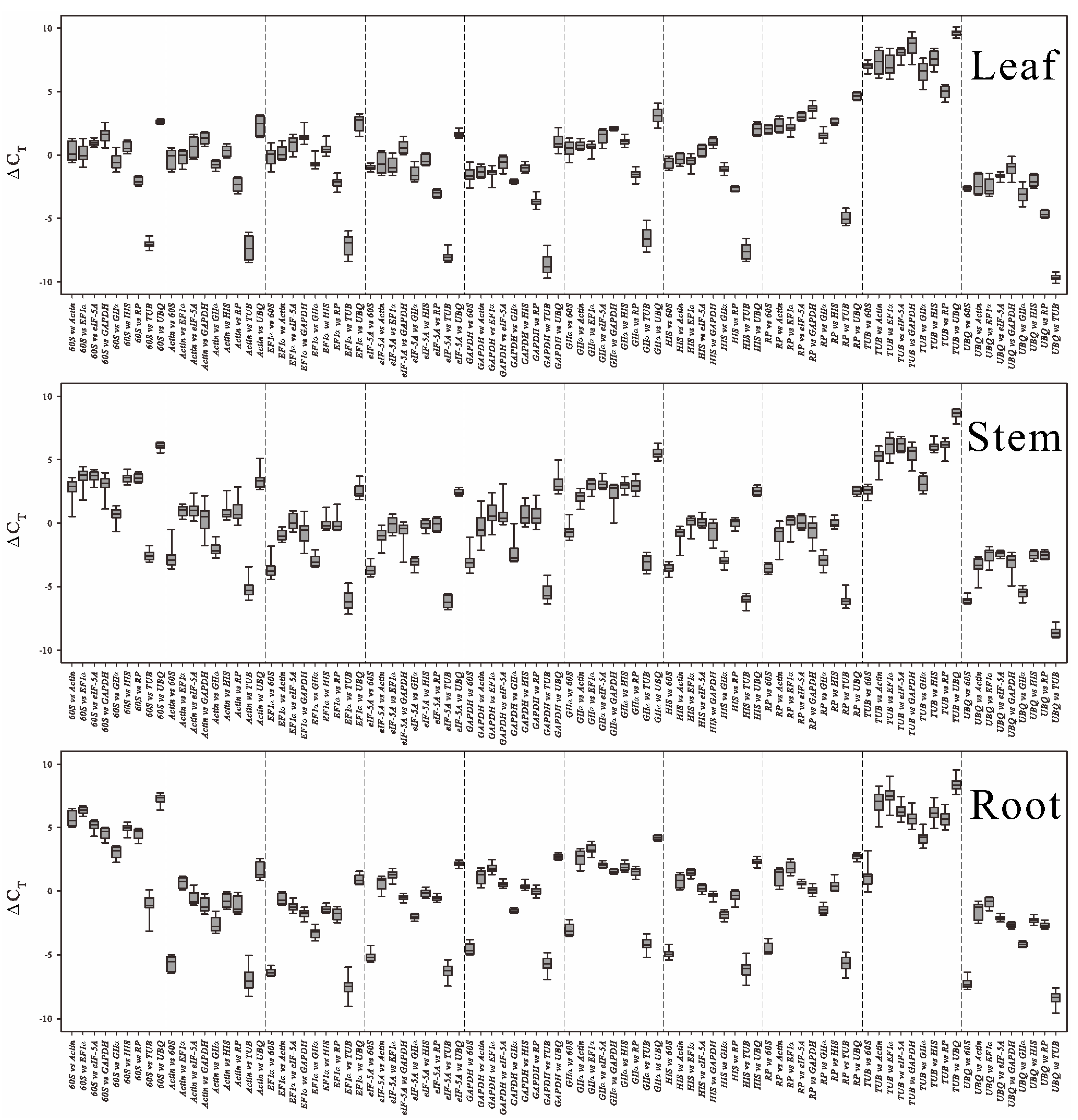
Figure 2 and
Figure S4). For example,
Actin had 3.22 cycles of variation in stem; if a target gene is normalized with
Actin or a 0.3 cycle variation reference gene, the final relative quantification will have a 2
3.22−0.3 = 7.57-fold discrepancy. This may fundamentally change the target gene expression profile. Thus, candidate genes showing high-level variation should be avoided as endogenous controls. More importantly, in our pre-experiments
18S did not have significant variation, and it is often used as a candidate reference gene, such as in two
Populus species for reference gene validation [
40,
51]. However, since the expression level of
18S was too high compared with other genes, the difference value was close to ten cycles, resulting in a 1024-fold discrepancy. To the best of our knowledge, most functional genes’ expression levels are less than that of
18S, and one of the principles in selecting suitable reference genes is that the expression level of the target and reference genes should be similar. For this reason,
18S is rarely used as a reference gene, so we omitted it in our study and did not take it as an appropriate candidate reference gene. To further analyze the expression stability of candidate genes, several algorithms, including geNorm [
11], NormFinder [
43], BestKeeper [
10], Δ
Ct [
42], qBasePlus [
52], RefFinder [
53], GenEx [
54], GrayNorm [
46], single-factor analysis of variance, and linear regression analysis [
51] have been applied. Among these algorithms, geNorm, and NormFinder have the greatest impact, and the latter one is superior. NormFinder and geNorm are free of charge. Meanwhile, geNorm has been integrated into the qBasePlus and GenEx tools, both of which are powerful RT-qPCR data analysis tools.
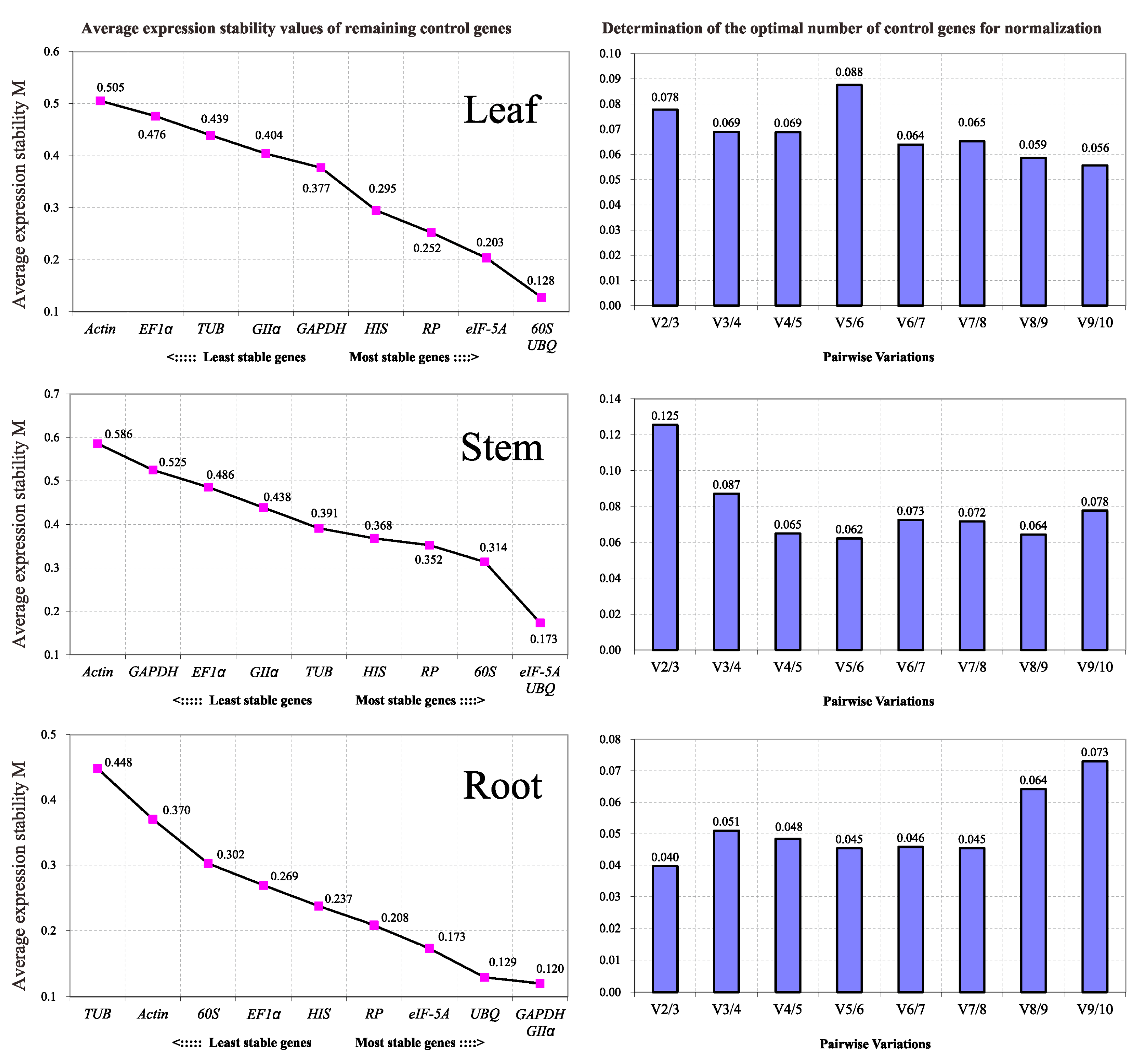
For our data analysis, we chose the ΔCt algorithm, the two most popular tools and a new algorithm, GrayNorm, to evaluate reference gene stability in P. euphratica. The ΔCt algorithm is a primary and relatively simple approach to rank gene stability based on the SD of pair-wise differences in Ct values. This uses only Cq values for calculation, while the other three tools introduce both Cq and efficiency values. In practice, PCR efficiency is an important factor that fluctuates along with coexisting substances originating from the RNA extract and cDNA synthesis, lengths and base sequences of the primers or amplification targets, quality of the Taq enzyme, and performance of the instrument. Although these factors are not considered in the ΔCt method, the simplicity of the algorithm makes it advantageous to use. The ΔCt method can be used for the initial screening of reference genes from a small number of candidates since it requires manual calculation, while the other three methods can perform automated calculations.
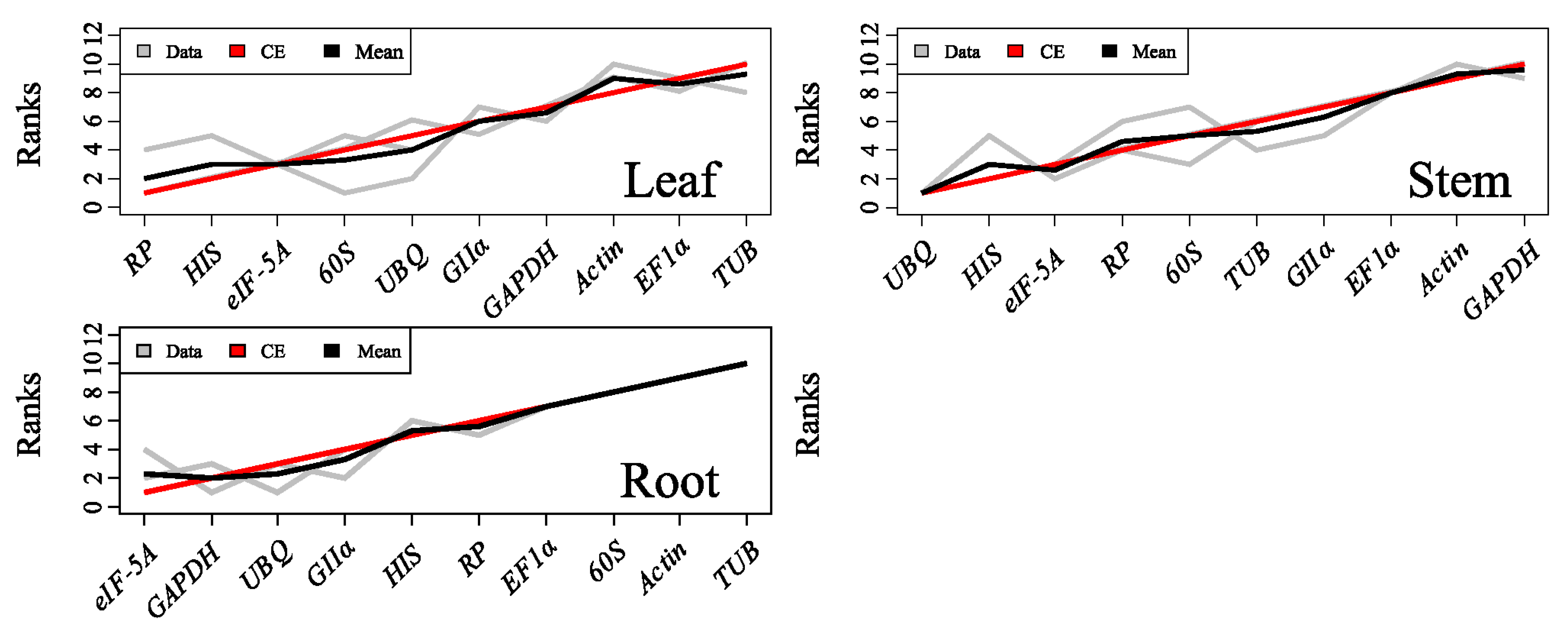
When determining the stability of the ten reference genes, the five methods mentioned above yielded different ranking patterns. Each method has its own algorithm, some of which are
Cq-based (Δ
Ct) while others are quantity-based (NormFinder, geNorm and GrayNorm). In addition, even methods with the same base algorithm showed discrepancies; for example, when transforming
Cq values to quantities to obtain input, NormFinder distributes the three
Cq value repeats to three groups, while geNorm uses the average
Cq values of the repeats, and GrayNorm provides all the gene combinations at one time. Meanwhile, because in all RT-qPCR analysis, GOI data are divided by
NF values during normalization, the algorithm of GrayNorm based on 1/
NF is more visualized. The closer the average 1/
NF per sample group to 1.0, the more stable of the reference gene [
46]. More importantly, GrayNorm displays all the gene combinations at one time from which the optimal combination can be easily found, so if only one algorithm is required, GrayNorm is a fine choice for RT-qPCR data analysis.
In previous studies on
Populus reference gene evaluation,
P. trichocarpa ×
Populus deltoids [
51] and
Populus × euramericana [
40], both experimental conditions are in different development stages, such as overwintered terminal vegetative buds, shoot tips including unexpanded leaves and terminal vegetative buds [
51], or adventitious rooting of
Populus hardwood cuttings [
40], neither of them is involved in abiotic stress. In our study, we focused on the extraordinary characteristic of
P. euphratica on salt stress resistance. The appropriate reference genes identified in our study were different with those in above mentioned
Populus.
UBQ,
ACT2 and
18S were the best three candidate reference genes in
P. trichocarpa × Populus deltoids,
EF1α and
18S were the optimal two ones in
Populus × euramericana. Meanwhile,
18S was the optimal reference gene in potato [
55], however, in our study,
18S was omitted because of its too-high expression level.
ACT2 (Actin) and
EF1α were middle-ranked, and only
UBQ was top-ranked in both
P. trichocarpa × Populus deltoids and
P. euphratica.
Moreover,
UBQ and
eIF-5A were two of the best choices for use in all tissues in our study, basically corresponding to the other species, such as rice [
56], soybean [
18],
Oryza sativa [
57], lettuce [
58] and
Arabidopsis [
59]. This may suggest that reference genes validated in
P. euphratica have commonality with other species, and these genes are conservative. Of even greater interest,
HIS can be considered as a novel reference gene appropriate for RT-qPCR in
P. euphratica. However,
Actin, widely used as endogenous control during plant development, should be avoided in
P. euphratica under salt stress.
In
P. euphratica DNA microarray,
GIIα have a constant expression in all experiments [
60], and this indicates that it can be regarded as an ideal reference gene. However, in our study,
GIIα ranks middle-level in all tissues. Its best ranks are fourth in both root and total, behind the widely used reference gene,
UBQ and
eIF-5A. Because the DNA microarray was focused on water deficit stress while our research conditions are salt stress, this indicates that in different experiment treatments, there are different optimal reference genes. As water deficit leads to osmotic stress while salt condition leads to both osmotic and ion stress [
61], maybe
GIIα is involved in
P. euphratica response to ion stress, and this may have an influence on its expression stability. For
GAPDH, it ranks seventhly in twenty-one candidate reference genes in
Arabidopsis (
GAPDH, AT1G13440) during development [
13], and shows middle-ranks in
Eucalyptus [
49], berry [
62], and tomato [
63] during different environment conditions, even in human and mouse cells [
64], and the validation results were similar with ours.
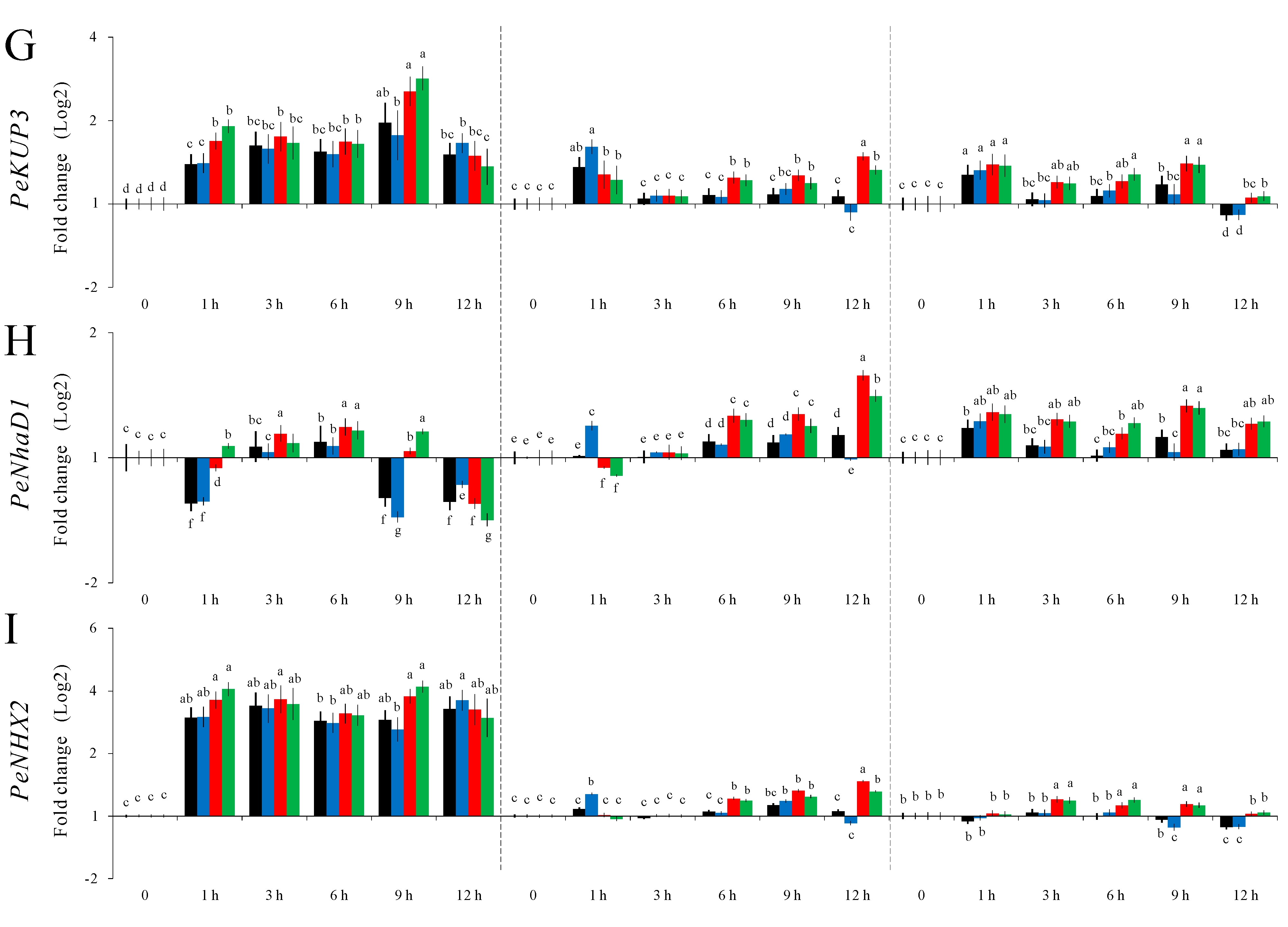
Most of the nine genes’ expression levels were salt-induced as seen in
Figure 6, indicating that they may participate in a salt-related mechanism in
P. euphratica. The salt-resistance contribution may be somewhat reflected by the transcript levels in plants.
PeNHX2, an NHX-type Na
+/H
+ exchanger gene, reported to play a role in mediating sodium tolerance in
P. euphratica [
65,
66], showed a much higher level (fold change) than the other eight genes in leaf, and this indicated that
PeNHX2 might have a principle role in maintaining ion balance under salt conditions. The results in
Figure 6 indicate that using different combinations of reference genes identified from various algorithms can influence target gene relative expression levels, and may lead to statistical significance. More importantly our results indicate that a combination of multiple reference genes recommended by GrayNorm algorithm, as shown in
Table 4, should be used instead of a single reference gene. Furthermore these identified functional genes can be applied in future research in understanding of
Populus salt-response signaling.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





