1. Introduction
Dendroctonus-bark beetles (Curculionidae: Scolytinae) play a fundamental role in the structure of coniferous forests, because they colonize and kill sick, damaged, and weakened trees, contributing to community succession, nutrient cycling, and canopy thinning [
1]. However, some of their species are also disturbance agents because, under certain forest stress conditions, outbreaks occur that affect hundreds of thousands of conifer trees of the genera
Larix,
Picea,
Pinus, and
Pseudotsuga from North and Central America [
2,
3,
4].
Dendroctonus-bark beetles are phytophagous insects that spend most of their life cycle under the bark of host plants, where they reproduce, feed on phloem, and interact with microbial symbionts and invertebrates. These symbionts have been isolated from the exoskeleton, galleries, and gut of these bark beetles [
5,
6,
7,
8,
9,
10]. A core gut bacteriome of
Dendroctonus species is known [
10], as well as the yeasts, mites, and nematodes diversity associated to them [
11]. Some of microbial symbionts are capable of degrading different substrates such as starch, esters, and lipids [
12], cellulose [
13], recycling uric acid [
7], and degrading or transforming different monoterpenes from the host trees of these insects [
14,
15,
16].
The phloem is a conduction system that transports different molecules and ions such as mRNAs, hormones, sugars, and nutrients such as trace elements and amino acids towards different plant body parts. As constitutive tissue is formed by different cell types (e.g., sieve cells and sieve tube elements integrating the conduction system itself, the companion and parenchyma cells), which represent a substrate rich in organic acids and non-structural carbohydrates (e.g., starch, sucrose, raffinose, stachyose, verbascose), as well as in structural carbohydrates (e.g., cellulose, hemicellulose). In particular, starch is the main carbohydrate reserve of the plants, synthesized from sugars produced during photosynthesis both in autotrophic and heterotrophic tissues (e.g., roots, woody tissues, fruits, seeds, tubers, and pollen grains) [
17].
Starch is stored in plants as insoluble particles or granules and is composed of amylose and amylopectin [
18]. Amylose constitutes about ~25% of starch and is essentially linear with α-D (1→4) linked glucose units and a few branched points per molecule. Amylopectin is highly branched and constitutes ~75% of starch, it is a polymer of α-D (1→4) linked glucose units and joined by α-D (1→6) linkages after every 24–30 glucose units [
19]. The structural complexity of starch requires different mechanisms for its hydrolysis, where enzymes such as α-amylases (α-1,4-glucan-4-glucanohydrolases; EC 3.2.1.1) are fundamental to catalyze this polysaccharide into low-molecular-weight molecules and other carbohydrates as well as α-dextrins, maltotriose, and maltose [
20,
21,
22]. Given that starch is the most abundant polysaccharide reserve in different plant tissues, it has been hypothesized that products derived from its hydrolysis might be utilized as essential food sources by insects for their development and survival. Several studies have reported that the number of α-amylase gene copies (from 1 to 13) is variable in insects. Some of them have been biochemically characterized, sequenced, and their phylogenetic relationship inferred, as well as their location, enzyme excretion sites, and regulatory mechanisms [
23]. Unfortunately, in bark beetles, α-amylases have received very little attention. Studies in population genetics have demonstrated the presence of allelic variants of these enzymes [
24]. However, Viktorinova et al. [
25] demonstrated in
I. typographus the presence of two α-amylase genes one of them producing two isoforms as a result of alternative splicing.
In this study, we analyzed the α-amylases of
Dendroctonus rhizophagus Thomas & Bright, an endemic species to the Sierra Madre Occidental in Mexico, which colonizes and kills seedlings and saplings (<3 m height and ~10 cm diameter) of several pine species [
26,
27]. The life cycle of
D. rhizophagus is annual, univoltine, and atypical among
Dendroctonus species. This species does not perform massive attacks on trees. Just one pair of insects colonizes and kills individual trees. In particular, we molecularly characterized the enzyme AmyDr, determined its number of isoforms, analyzed the relative expression of α-amylase
AmyDr gene through different developmental stages, provided functional evidence that both native and recombinant α-amylase AmyDr of this bark beetle are capable of hydrolyzing starch, and determined the effect of metal and non-metal ions on recombinant α-amylase activity.
3. Discussion
Our findings showed that AmyDr is a functional enzyme encoded by an ORF of 1452 nucleotides and is present both in the gut and head-pronotum of
Dendroctonus rhizophagus.
In silico analysis showed that this protein has a 51.51 kDa molecular mass (without signal peptide), similar to the α-amylase from other scolytines, such as
D. ponderosae (51.37 kDa, XP_019767850.1),
Ips typographus (51.84 kDa, ADQ54210.1; 52.19 kDa, ADQ54211.1), and
Hypothenemus hampei (51.24 kDa, AHY03307), curculionids, such as
Antohonomus grandis (50.87 kDa, AAN77138.1; 52.28 kDa, AAN77139.1) and
Sitophilus oryzae (51.32 kDa, ADM73187.1), other coleopterans, such as
Tenebrio molitor (51.24 kDa, P56634) and
Tribolium castaneum (51.60 kDa, AAA03708), and other insects, such as
Spodoptera frugiperda (54.78 kDa, AAO13754),
Drosophila melanogaster (51.93 kDa, AAA92232), and
Apis mellifera (54.25kDa, NP_001011598). The theoretical pI of AmyDr (4.54) is similar to the pI of coleopteran α-amylases (pI < 7) [
23].
Only one AmyDr isoform is present in the gut and head-pronotum of
D. rhizophagus, which agrees with the one found in other
Dendroctonus species such as
D. valens (MN782510),
D. frontalis (GAFI01015829.1), and
D. ponderosae (BT126692), and that reported in
H. hampei and other curculionids such as
Odoiporus longicollis (AHN92452.2) and
Cosmopolites sordidus (AKN63428.1). However, this result is different from those found in
A. grandis,
I. typographus, and
Rhynchophorus ferrugineus in which two
Amy genes have been documented [
25,
28,
29]. Based on the number of α-amylases genes (from 1 to 9) in Coleoptera [
23], as well as on the bioinformatics analyses of transcriptomes and genomes from
Dendroctonus species studied so far, the genus
Dendroctonus presented the lowest number of α-amylases genes.
The sequence analysis of AmyDr clones showed mainly synonymous substitutions along the sequences, suggesting the presence of allelic variants of the protein. In addition, transcriptome analyses of
D. rhizophagus, as well as from other
Dendroctonus species and scolytines such as
D. ponderosae (GAFW00000000),
D. frontalis (GAFI00000000),
D. valens [
30], and
Tomicus yunnanensis (GFJU00000000), referenced to
D. ponderosae genome, did not show evidence of isoforms. The absence of isoforms in
Dendroctonus-bark beetles suggests that the selection has favoured the occurrence of amylases with major functional plasticity, stability, and efficiency, but not the occurrence gene duplications. In contrast, in the bark beetle
I. typographus two functional α-amylases have been reported, one of them includes two isoforms [
25], an event rarely reported in animals because the common is the presence of multiple copies [
23] by gene duplications [
31] or horizontal transfer [
32]. In fact, a gene duplication event is evident in
Ips confusus transcripts (GIWS00000000), after our analysis developed on this project.
In other insects, it has been postulated that the existence of gene copies of α-amylase could be associated with an increase in their adaptive capacities to different food sources or to the occurrence of plant inhibitors [
33,
34]. Although the presence of inhibitors in the phloem pine trees has been documented,
Dendroctonus-bark beetles feed only on phloem. Stable and functionally generalist enzymes may be more efficient in an environment heterogeneous such as the subcortical region of the pine tree, which is degraded and changed as colonization proceeds and the tree dies.
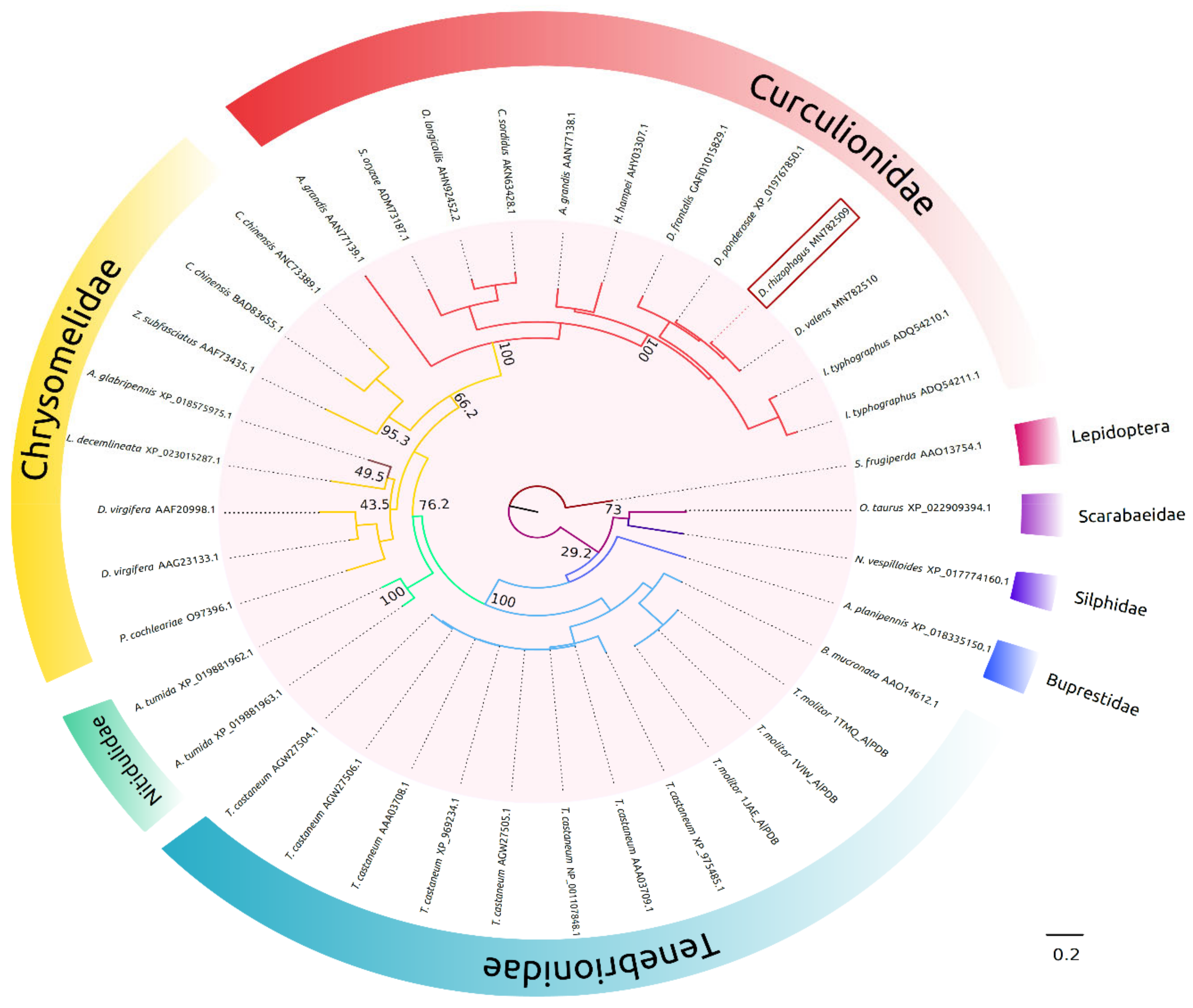
The phylogenetic analysis of AmyDr revealed the existence of a close relationship with the α-amylases of Curculionidae, and from these to those of Chrysomelidae, suggesting a common evolutionary history between both families of α-amylases. In addition, the presence of a short “flexible loop” with the absence of GHGA/GHG motif near the catalytic cleft in AmyDr and α-amylases from weevils and leaf beetles, suggests that this condition is conserved. Thus, like in other insect orders, two conditions exist in Coleoptera, namely one α-amylase group without motif and with a shorter “flexible loop” and the other group having a “flexible loop” with the presence of the GHGA/GHG motif [
31]. While both conditions have widely been documented, there are no convincing functional explanations or complete phylogenetic patterns that could enable us to hypothesize and explore why both conditions have evolved.
On the other hand, in contrast to both chlorine-dependent (arginine-bearing α-amylases) and chlorine-independent (glutamine-bearing α-amylases) α-amylases, AmyDr has a lysine (LYS319 without peptide-signal) in the third residue of the binding site to chlorine ion. This mutation is shared with other scolytines species (e.g.,
Dendroctonus,
Ips,
Hypotenemus), weevils (e.g.,
Anthonomus grandis), chrysomelids (e.g.,
Callosobruchus chinensis and
Zabrotes subfasciatus), and the two leaf beetle species analyzed (
Figure S1), as well as with the bacteria
Alteromonas haloplanktis [
35]. This mutation favours the union of ions NO
3− and Cl
−, which have been reported in other insect studies as weak activators. Our docking analyses showed that the binding of NO
3− to AmyDr (ARG182, ASN283, LYS319) is slightly stronger (binding energy −17.2 kJ mol
−1 and hydrogen bonds with distances of ≤2.2 Å) than that of Cl
− (−16.74 kJ mol
−1 and ≤2.1 Å), as previously demonstrated in marine bacterium
A. haloplanktis [
36].
Experimental results confirm the docking prediction because the activity of recombinant AmyDr is enhanced by the ions Ca
+2, and NO
3−, and slightly less by Cl
−. These findings indicate that AmyDr is conformationally stable and maintains its activity either in the presence or absence of these last two ions. It is known that insect α-amylases require calcium ions for the structural integrity and stability, and are activated by nitrates and chlorine ions. However, to our knowledge, there are no reports in other insects of lysine-bearing α-amylases or α-amylases with this mutation, functioning both in the absence and presence of these ions. The functional flexibility of α-amylase in bark beetles, as well as some weevils and phytophagous insects, suggests that this feature could be a functional adaptation associated with their habitat and feeding. In the case of
Dendroctonus bark beetles, this could be an adaptation to the changing subcortical environment where they develop and feed, which is apparently poor in calcium, nitrogen, and chlorine ions. These ions are acquired by trees through mineralization of the organic matter in the soil, where significant amounts of them are present [
37]. However, the nitrogen in bark beetles’ gut is mainly the result of the activity of their symbionts [
5,
38], and calcium and chlorine are acquired mainly through feeding but are actively regulated in the insect’s body by their functional role in different physiological processes.
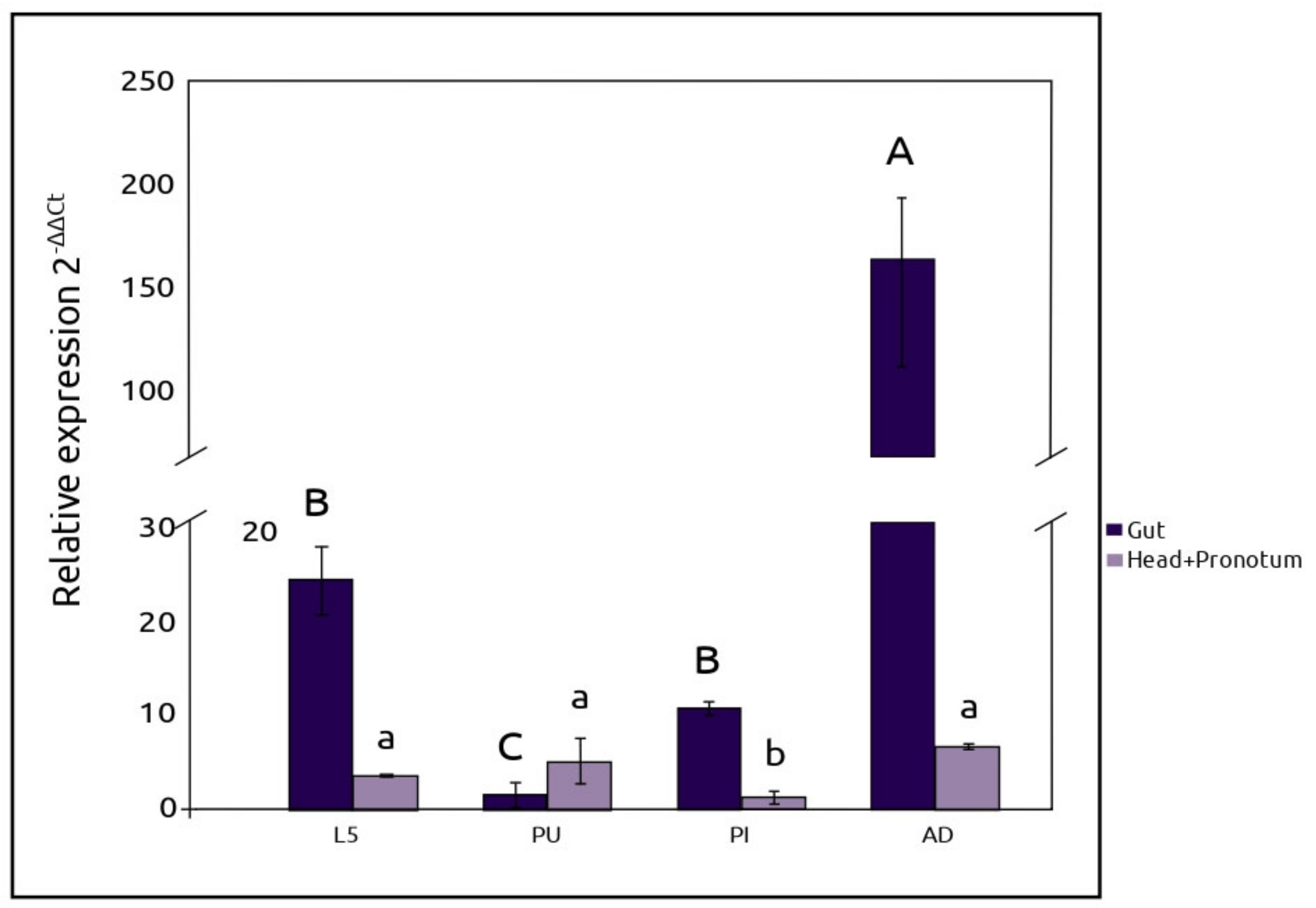
The observed expression of
AmyDr gene in the head-pronotum in different developmental stages, suggests that starch hydrolysis starts in the oral cavity of
D. rhizophagus. Whereas there is an obvious connection between starch hydrolysis and the nutrition process mainly in the midgut, amylolytic activity in the head-pronotum, salivary glands, and extraoral secretions have been frequently recorded in different insect species. Some authors have hypothesized that the lower amylolytic activity in the head-pronotum region, salivary glands, and extraoral secretions compared to the gut could be explained by the lack of optimal pH for the activity of α-amylases [
23,
39]. However, while this hypothesis may be applied to bark beetles, other factors, that might limit its activity, should be taken into consideration. For example, the high sclerotization of the anterior part of the bark beetles digestive system, which includes the oral cavity, pharynx, crop, and foregut, is structurally more associated to mechanic actions than digestive functions [
40,
41,
42]; the presence of inhibitors in the phloem cells that are released when the insect crushes the phloem [
34,
43]; and the strong energy demand that the synthesis of terpenoids represents, which are present in the induced resin that is produced once the insect colonizes the tree as part of the tree defence system [
44].
Lastly, the sharp decline in
AmyDr gene expression levels at the pupa and pre-imago stages compared to pre-emerged adults and larvae, indicates that the activity of the digestive processes in the gut is minimum at those stages. In addition, the general pattern of
AmyDr gene expression in this system suggests that the amylolytic activity is regulated through insect development. The different
AmyDr gene expression levels observed among the developmental stages of
D. rhizophagus can be explained by the processes of morphogenesis and organogenesis that occurs during the metamorphosis from larval-pupa, and pupa-pre-imago that limited food ingestion. On the other hand, we hypothesized that the regulation of amylolytic activity in the bark beetle may be associated with starch availability in the host tree. Several studies have reported that starch is accumulated in conifer needles, stem bark, and roots during spring [
45], just when adults of this bark beetle are found in the tree roots waiting to emerge in early summer with the start of the rain season [
46]. The growth and feeding of larval stages (I–IV) during summer and autumn matches with trees growth, when starch is hydrolyzed and its products moved to different parts of the tree to cover this energy cost. The fifth instar larvae finally migrate toward tree roots during early winter, where important starch reserves are stored. This hypothesis should be tested in further studies.
4. Materials and Methods
4.1. Insects
Naturally infested Arizona pines (Pinus arizonica Engelm) were collected at Mil Diez locality (23°48′28.12″ N 105°24′11.63″ W), Pueblo Nuevo municipality, Durango state, Mexico, from 2018 to 2019, and were transported immediately to the laboratory to get fifth-instar larvae (L5), pupa (PU), pre-imago (PI), and pre-emergent adult (AD) of D. rhizophagus. The gut was dorsally extracted and separated from the fat body and the Malpighian tubules; the head-pronotum and gut sections were separately placed into Eppendorf vials with 200 µL of Trizol® (Life Technologies, Carlsbad, CA, USA), and macerated completely using a sterile pestle. Three biological replicates of each stage were processed. As negative control, we used eggs collected directly from the galleries with fine-tip forceps. Three egg groups (n = 150) were integrated and placed in Eppendorf vials in the field, and several consecutive washes were done with distilled water and PBS buffer, and finally, they were stored in 200 µL of Trizol®. In the laboratory, they were completely macerated using a sterile pestle. For all the samples, vials were topped with 1 mL of Trizol® and stored at −80 °C for posterior RNA extraction and subsequent RT-qPCR.
4.2. DNA Cloning, Sequencing, and Full-Length Sequence Analyses
Total RNA was isolated from the whole adult body using the RiboPure™ RNA Purification kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. The cDNA was synthesized from 2 µg of total RNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA, USA) as per manufacturer’s protocol, and stored at −20 °C. Forward primer F1 5′ TGGCGATCAGTTCTAGACCAA 3′ and reverse primer R1 5′ ATTGCAACGTGCTCTCATAA 3′ were designed according to UTRs of a
D. rhizophagus’ putative α-amylase found in the transcriptome of this species [
30]. This sequence was similar to a putative α-amylase found in the genome of
D. ponderosae (XM_019912291.1) through Blastn, and to an α-amylase (AmyA, HQ417115) from the bark beetle
Ips typographus reported by [
25].
The PCR reaction was performed using Dream Taq Polymerase (Invitrogen, CA, USA) and the following conditions: 94 °C for 3 min, followed by 35 cycles of denaturation at 94 °C for 30 s, 57.5 °C annealing for 30 s, and extension at 72 °C for 2 min, with a final extension step at 72 °C for 20 min. PCR products were cloned into pGEM T Easy Vector (Promega, Madison, WI, USA) following the manufacturer’s instructions. A total of 30 clones with insert were sequenced in both senses at Macrogen, Inc. (Geumcheon-gu, Seoul, Korea).
The sequence obtained by PCR was confirmed by BlastX. To search different isoforms, the putative α-amylase sequence was mapped against all transcripts of adults and pupae transcriptomes from
D. rhizophagus [
30] using GMAP software, v. 2017-11-15 [
47] with a kmer of 12. An α-amylase encoding gene (
AmyDr) was deposited in the GenBank (accession number MN782509). The deduced amino acid sequence was obtained with the ExPasy Translate Tool (
http://www.expasy.org/tools/dna) and the Protein isoelectric point calculator (
http://isoelectric.org) was used to predict the physicochemical characteristics of the enzyme, including molecular mass (kDa) and isoelectric point (pI). Moreover, subcellular localization and signal peptide predictions were realized with the ProtComp-AN software, v.9.0 and SignalP 5.0 server (
http://www.cbs.dtu.dk/services/SignalP). The NetNGlyc v.1.0 (
http://www.cbs.dtu.dk/services/NetNGlyc) and NetOGlyc v.4.0 (
http://www.cbs.dtu.dk/services/NetOGlyc) servers were used to predict potential N and O glycosylation sites.
4.3. Phylogenetic Analysis
Multiple protein alignment with reference coleopteran sequences was performed with Muscle software, v.3.8.31 [
48] and a phylogenetic inference analysis by maximum likelihood was carried out in PhyML 3.0 server (
http://www.atgc-montpellier.fr/phyml/). The more appropriate model of protein evolution was determined using the Akaike information criteria (AIC = 25857.30, − lnL = −12834.65) and Bayesian information criteria (BIC = 26253.66, − lnL = −12834.65) [
49]. Both criteria supported the model LG + G + I + F [
50], and the support of nodes was evaluated after 1000 bootstrap pseudoreplicates. The α-amylase sequence of
Spodoptera frugiperda (AAO13754.1) was used as outgroup.
4.4. Molecular Modelling and Docking
The 3D structure of the α-amylase was obtained by homology modelling with Modeller software, v.9.16 [
51], using as a template the α-amylase crystalized structure obtained by the X-Ray of
Tenebrio molitor (PDB ID: 1TMQ, 1JAE), which is the only structural model of α-amylases in insects elucidated by X-Ray [
52]. The best model was selected and validated in the Structural Analysis and Verification Server (
http://servicesn.mbi.ucla.edu/SAVES) using Verify3D [
53], Errat [
54], Procheck [
55], and Whatcheck [
56]. The Ramachandran plot was generated [
57] in the RAMPAGE server (
http://mordred.bioc.cam.ac.uk/~rapper/rampage.php). To identify active and binding sites with calcium and chlorine ions, a multiple alignment without signal peptides of AmyDr sequence from
D. rhizophagus and several α-amylases from coleopterans and lepidopterans was performed in ESPript software, v.3.0 [
58]. The 3D structure of the α-amylase of this bark beetle was analyzed using the PyMol™ software, v.2.2.0 [
59].
Predicted interactions between amylopectin and amylose with ions were evaluated using docking analyses. Ligands were drawn in ChemDraw software, v.15.0 (
http://www.cambridgesoft.com) and were geometrically optimized with the Gaussian software, v.5.0.9 [
60] using AM1 and DFT (B3LYP/6-31G(d,p)) levels. The output files were converted to .pdb files, and the ligand files were viewed using the AutoDockTools (ADT software), v.4.2 [
61]. To perform docking analyses, the Kollman charges for all atoms were computed prior to the addition of polar hydrogens to the α-amylase, and torsion angles in the small flexible ligands were included in the Monte Carlo algorithm. The protein exploration and definition of the binding site were prepared using a GRID-based procedure [
62]. All docking simulations employed the hybrid Lamarckian genetic algorithm with an initial population of 100 randomly placed individuals and 1 × 10
7 energy evaluations. The interactions between the ligands and the α-amylase were visualized using ADT software, v.4.2, and the figures were created using the PyMol™ software, v.2.2.0.
4.5. Amylolytic Activity and Peptide Identification
Ten adult beetles of D. rhizophagus (1:1 sex ratio) were placed in holes drilled (0.5 cm) into the fresh stumps of Arizona pines (40 cm high and ~15 cm diameter) and secured with steel mesh and staples. The beetles were fed for 12 h at room temperature in dark conditions. Then, the head-pronotum was removed directly from the insects, and the guts were dorsally extracted after the elytra and wings. The head-pronotum and gut from insects were separately homogenized in 100 µL 1 mM phenylmethylsulfonyl fluoride (PMSF) and centrifuged at 10,000× g for 15 min at 4 °C. The pellets of each sample were measured, normalized, and loaded by duplicate with the sample buffer (0.05 M Tris HCl, pH 7.0; 20% glycerol, 0.05% Triton X-100; 0.01% bromophenol blue) and incubated at 37 °C for 10 min. The samples were loaded twice into an 8% polyacrylamide gel copolymerized with 1% (w/v) of starch (Sigma-Aldrich, St. Louis, MO, USA) using a Mini PROTEAN II system with a running buffer free of SDS at pH 8.3. Native-PAGE was conducted at 4 °C for 4 h at 60 V. After electrophoresis, the gel was rinsed with distilled water for 15 min, and the zymogram assay was performed in 50 mM citrate buffer, pH 5.0, for 1 h at 40 °C.
The gel was cut into two longitudinal sections. The first section was stained with Lugol’s solution for 5 min and incubated in distilled water with shaking to visualize the amylolytic activity. The second section was not stained but was matched with the first section once activity was visualized. To determinate the presence of α-amylase peptides from the head-pronotum and gut samples using MALDI-TOF/TOF, the lane was divided into four parts (S1–S4). Due to amylolytic activity displayed in the section S3 in both samples, it was divided into two subsections (a, b) for the gut sample and (a’, b’) for the head-pronotum sample. These sections were cut and digested with trypsin to identify peptides according to Shevchenko et al. [
63]. The resulting peptides were concentrated in an approximate volume of 10 μL. Thereafter, 9 μL was desalted using C18 Zip-Tips (Millipore, Burlington, MA, USA). ChromXP Trap Column C18-CL precolumn (Eksigent, Dublin, CA, USA); 350 μm X 0.5 mm, 120 Å pore size, 3 μm particle size and desalted with 0.1% trifluoroacetic acid (TFA) in H
2O at a flow rate of 5 μL min
-1 during 10 min. Then, peptides were loaded and separated on a 3C18-CL-120 column (Eksigent, Dublin, CA, USA); 75 μm X 150 mm, 120 Å pore size, 3 μm particle size, in an HPLC Ekspert nanoLC 425 (Eksigent, Dublin, CA, USA) using as mobile phase A, 0.1% TFA in H
2O, and mobile phase B 0.1% TFA in acetonitrile (ACN) under the following linear gradient: 0-3 min 10% B, 60 min 60% B, 61–64 min 90% B, 65 to 90 min 10% B at a flow rate of 250 µL min
−1. Eluted fractions were automatically mixed with a solution of 2 mg mL
−1 of α-cyano-4-hydroxycinnamic acid in 0.1% TFA and 50% ACN as a matrix, spotted in an Opti-TOF plate of 384 spots using a MALDI Ekspot (Eksigent, Dublin, CA, USA) with a spotting velocity of 20 s per spot at a matrix flow rate of 1.6 μL min
−1. The spots generated were analyzed by a MALDI-TOF/TOF 4800 Plus mass spectrometer (ABSciex, Framingham, MA, USA). Each MS Spectrum was acquired by an accumulation of 1000 shots in a mass range of 850–4000 Th with a laser intensity of 3800. The 100 more intense ions with a minimum signal-noise of 20 were programmed to fragmenting. The MS/MS spectrums were obtained by the fragmentation of selected precursor ions using collision-induced dissociation and acquired by 3000 shots with a laser intensity of 4300. Generated MS/MS spectrums were compared against the Swiss-Prot database by the Protein Pilot software, v.2.0.1, using the Paragon algorithm. Search parameters were: Carbamidomethylated cysteine, trypsin as a cut enzyme, all the biological modifications and amino acid substitution set by the algorithm; as well as phosphorylation emphasis, and Gel-based ID as special factors. The detection threshold was considered in 1.3 to acquire 95% of confidence. Also, the identified proteins observed a local false discovery rate of ≤ 5%. In a bid to minimize redundancy, identified proteins were grouped by the ProGroup algorithm in Protein Pilot software, v.2.0.1. Analyses were carried out by the “Unidad de Genómica, Proteómica y Metabolómica” at LaNSE, CINVESTAV, CDMX, México.
4.6. RT-qPCR
The relative expression of the α-amylase was evaluated in the different developmental stages of the
D. rhizophagus life cycle (L5, PU, PI, and AD) and the different tissues (head-pronotum and gut). Total RNA from the samples was isolated using the RiboPure™ RNA Purification kit (Life Technologies, Carlsbad, CA, USA). The RNA concentration and purity (A
260/A
280 ratio) were determined using an ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and its integrity was checked in 1% agarose gel electrophoresis. The cDNA synthesis was done using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) from 2 μg total RNA in a final reaction volume of 20 μL, as per manufacturer’s protocol. TaqMan
® probes and primers were designed by Applied Biosystems (
Table S1). The TaqMan
® hydrolysis probes were labeled at the 5′ end with the reporter dye 6-carboxyfluorescein (FAM) and joined at the 3’ end to a non-fluorescent quencher molecule group MGB (Minor Groove Binder). The reactions were carried out with a Step One
® TM Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) in a final volume of 20 μL: 20X primer and probes with a final concentration of 900 nM of each primer, 250 nM of the TaqMan probe, TaqMan
® Universal Master Mix II (Applied Biosystems, Carlsbad, CA, USA) and 5 μL of 100-fold dilution of cDNA. All samples were placed in Fast Optical 48-well reaction plates MicroAmp
®™ (Thermo Fisher Scientific, Carlsbad, CA, USA). Standard manufacturer’s amplification conditions were used: 50 °C for 2 min, 95 °C for 10 min, 40 cycles at 95 °C for 15 s, and 60 °C for 60 s. PCR contamination was undetected in the no template control NTCs (unintended amplification products) and three technical replicates were performed for each biological replicate from each development state. A set of ten guts or ten head-pronotum were used in each of the replicas.
Glyceraldehyde 3-phosphate dehydrogenase (
GAPDH; MN782508) was used as a reference gene since it showed a stable expression according to pupa and adult transcriptomes of
D. rhizophagus [
30]. The RT-qPCR efficiency and validation for the reference gene were assessed using a linear regression analysis with average values obtained in three replicates of the quantification cycles (Ct) with five cDNA dilutions (an initial 1:20 dilution and four subsequent 1:5 dilutions). The PCR efficiency for
GAPDH and
Amy genes were estimated with the relation: efficiency = (10
−1/slope−1)100, where the slope values were −3.58 and −3.52 respectively, and R
2 value of 0.99. To evaluate the specificity, all amplicons were visualized in 1% agarose gel electrophoresis. All experimental procedures were performed according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (
Supplementary Table S2) [
64].
4.7. Statistical Analysis
The relative expression values of the α-amylase gene were determined using the 2
−ΔΔCt method [
65], where the Ct values of the egg stage and the
GAPDH gene were used for data normalization. To evaluate significant differences in the expression level of the α-amylase gene, a one-way ANOVA with Welch’s
F and Tukey test were performed for the two different tissues (head-pronotum and gut) and different developmental stages (fifth-instar larvae, pupa, pre-imago, and pre-emergent adult). Statistical tests were performed with Past software, v.3.26 [
66]. Values of
p < 0.05 were considered significant.
4.8. Expression of Recombinant AmyDr
Plasmid with recombinant AmyDr was recovered from DH5α cells with the construction pGEM_AmyDr, using the GenElute TM Plasmid Midiprep kit (Sigma-Aldrich Corp., St. Louis, MO, USA) following manufacturer’s protocol. The AmyDr open reading frame (ORF) was PCR-amplified with primers F1_DR1_SPIF (5′GTCGACTGGATCCGGATGCAGCTAGGAATTCTG3′) and R1_DR1_SPIF (5′ATCTCGAGTGCGGCCTTATAATTTTGCATTTACATG3′) using a Q5 High-Fidelity DNA Polymerase (New England BioLabs, Inc., Ipswigh, MA, USA) following manufacturer’s protocol. The amplification product was cloned into pENTR4 using In-Fusion HD EcoDry (Takara Bio, inc., Mountain View, CA, USA) and transformed into Stellar™ Competent Cells (E. coli HST08) (Takara Bio, inc., Mountain View, CA, USA) following the manufacter’s instructions. The recombinant plasmid was confirmed by sequencing prior recombination into the linearized BaculoDirect™ C-term vector (Invitrogen, Carlsbad, CA, USA) by LR recombination with LR Clonase™ II (Invitrogen, Carlsbad, CA, USA). Sf9 cells were infected with the recombinant Bac_AmyDr vector to obtain a high-titer P3 viral stock by successive amplifications of P1 and P2 stocks following manufacturer’s protocol.
Recombinant AmyDr P3 viral stock (500 μL), previously produced, was used to infect 1 × 106 Sf9 cells mL−1 in 30 mL of Sf900 II serum-free media (Gibco, Grand Island, NY, USA) suspension cultures for 72 h at 27 °C. Cells expressing recombinant AmyDr were harvested with Cell Lysis Buffer (100 mM sodium phosphate, pH 7.6, 20% (vol/vol) glycerol, and 1.1 mM EDTA) supplemented with 75 μL of phenylmethylsulfonyl fluoride (0.1 M PMSF), 15 μL of protease inhibitor cocktail (PIC) (Sigma-Aldrich Corp., Milwaukee, WI, USA), and 30 μL of dithiothreitol (0.1 M DTT), and then centrifuged at 3000× g at 4 °C for 10 min. The supernatant was isolated and tested for functional AmyDr. As negative control of amylases expression, 30 mL of 1 × 106 Sf9 cells mL−1 in Sf-900 II SFM suspension culture without viral stock and processed as mentioned before was used.
4.9. Preparation of Amylase Crude Extract
The total protein from AmyDr crude extract and the negative control from Sf9 cells without viral stock were precipitated overnight at 4 °C and 80% (NH4)2SO4 saturation. The pellet was recovered by centrifugation at 8800× g at 4 °C for 20 min, suspended in 5 mL of 50 mM, pH 5.0 citrate buffer, and dialyzed at 4 °C for 12 h in a cellulose membrane (Spectrum, Zarauz, Guipúzcoa, Spain) using the same buffer. Proteins obtained were isolated in 10% SDS-PAGE.
4.10. Protein Electrophoresis
Protein analyses were performed in 10% SDS-PAGE using a Mini PROTEAN II system. Proteins in the gel were visualized using Coomassie Brilliant Blue R-250 and recorded with a MiniBIS Pro documentation system (DNR Bio-Imaging Systems Ltd., Jerusalem, Israel). Protein molecular weight (MW) was estimated using as reference broad-range molecular weight protein standards (Thermo Fisher Scientific, Waltham, MA, USA).
4.11. Enzyme Assay
The amylolytic activity of AmyDr was determined on 1% starch agar plates (10 g L−1 starch, 23 g L−1 agar dissolved in deionized water), where 50 μL of amylase crude extract were added in a 5 mm hole, previously made with a Pasteur pipette on starch agar plates. These were incubated at 37 °C for 12h and stained with Lugol’s solution to document the enzyme activity around the hole.
4.12. Effect of Metal and Non-Metal Anions on Enzyme Activity
To evaluate the effect of metal ions, such as Ca2+ and Cl−, and non-metal such as NO3− on the enzymatic activity of AmyDr, samples of the crude extract were incubated in CaCl2, NH4Cl, Ca(NO3)2, and KNO3 at two final concentrations of 1 and 5 mM each. Likewise, as described before, the reaction mixture was added in a 5-mm hole on the starch agar plates, incubated at 37 °C for 12 h, and stained with Lugol’s solution to document the enzyme activity around the hole. Amylolytic activity was compared to a control plate with only metal and non-metal ions, and a control plate with Sf9 cells including metal and non-metal ions, which were the negative control of this assay.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







