
Genetic Aspects of Age-Related Macular Degeneration and Their Therapeutic Potential
 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
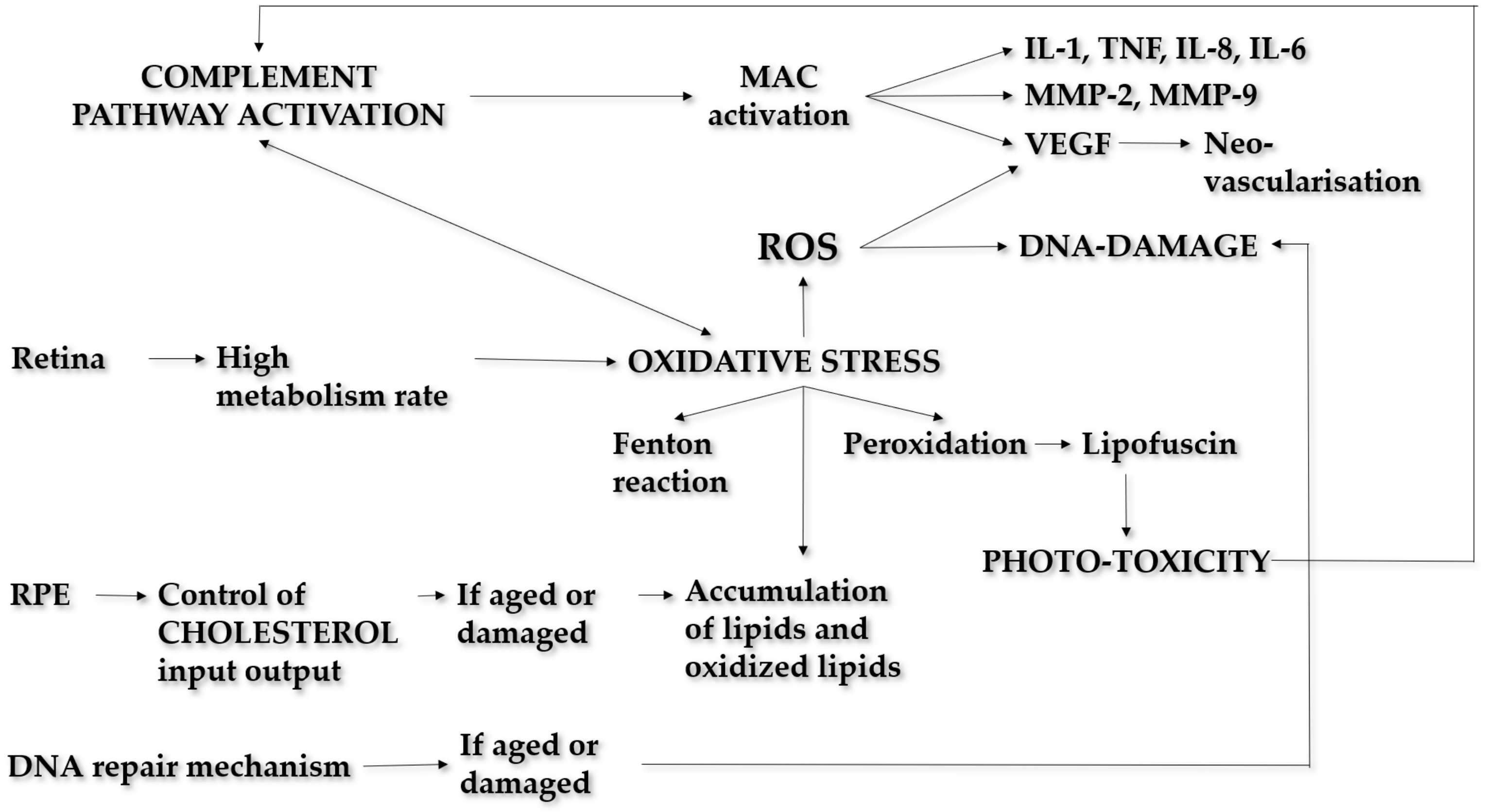
Pathogenesis
2. Methods
3. Results
3.1. AMD Genomics
3.1.1. Immune Response and Complement Genes
Complement Factor H (CFH)
Complement-Factor-H-Related (CFHR)
Complement C3
Complement Factor B/Complement 2 (CFB/C2)
Complement C9
Complement Factor I (CFI)
Other Genetic Variants
3.1.2. PLEKHA/ARMS2/HTRA-1
3.1.3. Oxidative Stress Genes
Manganese Superoxide Dismutase (MnSOD)
Iron Homeostasis Genes
3.1.4. Lipid Metabolism Genes
Apolipoproteins (Apo)
ATP-Binding Cassette Family: ABCA1 and ABCA4
Hepatic Lipase (LIPC)
3.1.5. Cell Survival Genes
DNA Damage Repairing
Nuclear Factor Kappa B (NF-κb)
3.1.6. Neovascularisation
Vascular Endothelial Growth Factor (VEGF)
Tissue Inhibitor of Matrix Metalloproteinase 3 (TIMP3)
3.1.7. Extracellular Matrix Alterations
3.2. Clinical Applications
3.2.1. Association with Particular Clinical Subtypes of AMD
3.2.2. Detection of Bio-Markers
3.2.3. Genetic Tests
Prediction of Risk Profile
Response to Therapy
3.3. Future Perspectives
3.3.1. Development of Personalized Treatments
Complement
3.3.2. Gene Therapy in AMD
Anti-VEGF
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Adewoyin, T.; Chong, N.v. Age-related macular degeneration: A perspective on genetic studies. Eye 2008, 22, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Fine, S.L.; Berger, J.W.; Maguire, M.G.; Ho, A.C. Age-Related Macular Degeneration. N. Engl. J. Med. 2000, 342, 483–492. [Google Scholar] [CrossRef]
- Ferris, F.L.; Davis, M.D.; Clemons, T.E.; Lee, L.Y.; Chew, E.Y.; Lindblad, A.S.; Milton, R.C.; Bressler, S.B.; Klein, R. A simplified severity scale for age-related macular degeneration: AREDS Report No. 18. Arch. Ophthalmol. 2005, 123, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.F.; Dewald, A.D.; Streb, L.M.; Wang, K.; Kuehn, M.H.; Stone, E.M. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp. Eye Res. 2011, 93, 565–567. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Singh, R. The role of complement membrane attack complex in dry and wet AMD—From hypothesis to clinical trials. Exp. Eye Res. 2019, 184, 266–277. [Google Scholar] [CrossRef]
- Lueck, K.; Wasmuth, S.; Williams, J.; Hughes, T.R.; Morgan, B.P.; Lommatzsch, A.; Greenwood, J.; Moss, S.E.; Pauleikhoff, D. Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye 2011, 25, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Joly, S.; Francke, M.; Ulbricht, E.; Beck, S.; Seeliger, M.; Hirrlinger, P.; Hirrlinger, J.; Lang, K.S.; Zinkernagel, M.; Odermatt, B.; et al. Cooperative phagocytes: Resident microglia and bone marrow immigrants remove dead photoreceptors in retinal lesions. Am. J. Pathol. 2009, 174, 2310–2323. [Google Scholar] [CrossRef]
- Cruz-Guilloty, F.; Saeed, A.M.; Duffort, S.; Cano, M.; Ebrahimi, K.B.; Ballmick, A.; Tan, Y.; Wang, H.; Laird, J.M.; Salomon, R.G.; et al. T cells and macrophages responding to oxidative damage cooperate in pathogenesis of a mouse model of age-related macular degeneration. PLoS ONE 2014, 9, e88201. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Blasiak, J.; Synowiec, E.; Salminen, A.; Kaarniranta, K. Genetic variability in DNA repair proteins in age-related macular degeneration. Int. J. Mol. Sci. 2012, 13, 13378–13397. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.; Szaflik, J.P.; Zaras, M.; Sklodowska, A.; Janik-Papis, K.; Poplawski, T.R.; Blasiak, J.; Szaflik, J. DNA damage/repair and polymorphism of the hOGG1 gene in lymphocytes of AMD patients. J. Biomed. Biotechnol. 2009, 2009, 827562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, A.; ** rare, deleterious mutations in Factor H: Association with early onset, drusen burden, and lower antigenic levels in familial AMD. Sci. Rep. 2016, 6, 31531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, D.; Seddon, J.M. Phenotypic Characterization of Complement Factor H R1210C Rare Genetic Variant in Age-Related Macular Degeneration. JAMA Ophthalmol. 2015, 133, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Duvvari, M.R.; Saksens, N.T.; van de Ven, J.P.; de Jong-Hesse, Y.; Schick, T.; Nillesen, W.M.; Fauser, S.; Hoefsloot, L.H.; Hoyng, C.B.; de Jong, E.K.; et al. Analysis of rare variants in the CFH gene in patients with the cuticular drusen subtype of age-related macular degeneration. Mol. Vis. 2015, 21, 285–292. Available online: https://pubmed.ncbi.nlm.nih.gov/25814826 (accessed on 15 March 2015).
- Fritsche, L.G.; Fleckenstein, M.; Fiebig, B.S.; Schmitz-Valckenberg, S.; Bindewald-Wittich, A.; Keilhauer, C.N.; Renner, A.B.; Mackensen, F.; Mößner, A.; Pauleikhoff, D.; et al. A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Warwick, A.; Gibson, J.; Sood, R.; Lotery, A. A rare penetrant TIMP3 mutation confers relatively late onset choroidal neovascularisation which can mimic age-related macular degeneration. Eye 2016, 30, 488–491. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Braun, T.A.; Russell, S.R.; Kuehn, M.H.; Lotery, A.J.; Moore, P.A.; Eastman, C.G.; Casavant, T.L.; Sheffield, V.C. Missense Variations in the Fibulin 5 Gene and Age-Related Macular Degeneration. N. Engl. J. Med. 2004, 351, 346–353. [Google Scholar] [CrossRef]
- Kucukevcilioglu, M.; Patel, C.B.; Stone, E.M.; Russell, S.R. Clinically detectable drusen domains in fibulin-5-associated age-related macular degeneration (AMD). Int. Ophthalmol. 2016, 36, 569–575. [Google Scholar] [CrossRef]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.; Yu, Y.; Schramm, E.C.; Triebwasser, M.; Wagner, E.K.; Raychaudhuri, S.; Daly, M.J.; Atkinson, J.P.; Seddon, J.M. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum. Mol. Genet. 2015, 24, 3861–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.; Hartnett, M.E.; Atkinson, J.P.; Giclas, P.C.; Rosner, B.; Seddon, J.M. Plasma complement components and activation fragments: Associations with age-related macular degeneration genotypes and phenotypes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.S.; Teixeira, A.G.; Bavia, L.; Lin, F.; Velletri, R.; Belfort, R., Jr.; Isaac, L. Plasma levels of complement proteins from the alternative pathway in patients with age-related macular degeneration are independent of Complement Factor H Tyr402His polymorphism. Mol. Vis. 2012, 18, 2288–2299. Available online: https://pubmed.ncbi.nlm.nih.gov/22969267 (accessed on 30 August 2012).
- Stanton, C.M.; Yates, J.R.; den Hollander, A.I.; Seddon, J.M.; Swaroop, A.; Stambolian, D.; Fauser, S.; Hoyng, C.; Yu, Y.; Atsuhiro, K.; et al. Complement factor D in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8828–8834. [Google Scholar] [CrossRef]
- Scholl, H.P.; Charbel Issa, P.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T.; et al. Systemic complement activation in age-related macular degeneration. PLoS ONE 2008, 3, e2593. [Google Scholar] [CrossRef]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Charbel Issa, P.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2009, 19, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Tsai, D.C.; Charng, M.J.; Lee, F.L.; Hsu, W.M.; Chen, S.J. Different plasma levels of vascular endothelial growth factor and nitric oxide between patients with choroidal and retinal neovascularization. Ophthalmologica 2006, 220, 246–251. [Google Scholar] [CrossRef]
- Paun, C.C.; Ersoy, L.; Schick, T.; Groenewoud, J.M.; Lechanteur, Y.T.; Fauser, S.; Hoyng, C.B.; de Jong, E.K.; den Hollander, A.I. Genetic variants and systemic complement activation levels are associated with serum lipoprotein levels in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7766–7773. [Google Scholar] [CrossRef]
- Acar, İ.E.; Lores-Motta, L.; Colijn, J.M.; Meester-Smoor, M.A.; Verzijden, T.; Cougnard-Gregoire, A.; Ajana, S.; Merle, B.M.J.; de Breuk, A.; Heesterbeek, T.J.; et al. Integrating Metabolomics, Genomics, and Disease Pathways in Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2020, 127, 1693–1709. [Google Scholar] [CrossRef]
- Morohoshi, K.; Patel, N.; Ohbayashi, M.; Chong, V.; Grossniklaus, H.E.; Bird, A.C.; Ono, S.J. Serum autoantibody biomarkers for age-related macular degeneration and possible regulators of neovascularization. Exp. Mol. Pathol. 2012, 92, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.M.; Proia, A.D.; Johnson, W.H.; Cyr, D.; Lashkari, K. Interferon γ-inducible protein-10 (IP-10) and eotaxin as biomarkers in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, H.; Mamalis, C.; McFadden, M.; Taggart, M.; Stagg, B.; Passi, S.; Earle, P.; Chakravarthy, U.; Hogg, R.E.; Ambati, B.K. The reduction of serum soluble Flt-1 in patients with neovascular age-related macular degeneration. Am. J. Ophthalmol. 2015, 159, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stravalaci, M.; Ferrara, M.; Pathak, V.; Davi, F.; Bottazzi, B.; Mantovani, A.; Medina, R.J.; Romano, M.R.; Inforzato, A. The Long Pentraxin PTX3 as a New Biomarker and Pharmacological Target in Age-Related Macular Degeneration and Diabetic Retinopathy. Front. Pharmacol. 2022, 12, 811344. [Google Scholar] [CrossRef]
- Stravalaci, M.; Davi, F.; Parente, R.; Gobbi, M.; Bottazzi, B.; Mantovani, A.; Day, A.J.; Clark, S.J.; Romano, M.R.; Inforzato, A. Control of Complement Activation by the Long Pentraxin PTX3: Implications in Age-Related Macular Degeneration. Front. Pharmacol. 2020, 11, 591908. [Google Scholar] [CrossRef]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin. Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.J.; Verma, V.; Rosenberg, K.I.; Chan, C.C.; Tuo, J. Genetic markers and biomarkers for age-related macular degeneration. Expert. Rev. Ophthalmol. 2007, 2, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Qassim, A.; Souzeau, E.; Hollitt, G.; Hassall, M.M.; Siggs, O.M.; Craig, J.E. Risk Stratification and Clinical Utility of Polygenic Risk Scores in Ophthalmology. Transl. Vis. Sci. Technol. 2021, 10, 14. [Google Scholar] [CrossRef]
- Chatterjee, N.; Shi, J.; García-Closas, M. Develo** and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- Jakobsdottir, J.; Gorin, M.B.; Conley, Y.P.; Ferrell, R.E.; Weeks, D.E. Interpretation of genetic association studies: Markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet. 2009, 5, e1000337. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Liu, Y.; Yan, Q.; Fritsche, L.G.; Cook, R.J.; Clemons, T.; Ratnapriya, R.; Klein, M.L.; Abecasis, G.R.; Swaroop, A.; et al. Bivariate Analysis of Age-Related Macular Degeneration Progression Using Genetic Risk Scores. Genetics 2017, 206, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitendijk, G.H.S.; Rochtchina, E.; Myers, C.; van Duijn, C.M.; Lee, K.E.; Klein, B.E.K.; Meuer, S.M.; de Jong, P.T.V.M.; Holliday, E.G.; Tan, A.G.; et al. Prediction of age-related macular degeneration in the general population: The Three Continent AMD Consortium. Ophthalmology 2013, 120, 2644–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Rosner, B. Validated Prediction Models for Macular Degeneration Progression and Predictors of Visual Acuity Loss Identify High-Risk Individuals. Am. J. Ophthalmol. 2019, 198, 223–261. [Google Scholar] [CrossRef] [PubMed]
- Joachim, N.; Kifley, A.; Colijn, J.M.; Lee, K.E.; Buitendijk, G.H.S.; Klein, B.E.K.; Myers, C.; Meuer, S.M.; Tan, A.G.; Flood, V.; et al. Joint Contribution of Genetic Susceptibility and Modifiable Factors to the Progression of Age-Related Macular Degeneration over 10 Years: The Three Continent AMD Consortium Report. Ophthalmol. Retina 2018, 2, 684–693. [Google Scholar] [CrossRef]
- Stone, E.M.; Aldave, A.J.; Drack, A.V.; Maccumber, M.W.; Sheffield, V.C.; Traboulsi, E.; Weleber, R.G. Recommendations for Genetic Testing of Inherited Eye Diseases: Report of the American Academy of Ophthalmology Task Force on Genetic Testing. Ophthalmology 2012, 119, 2408–2410. [Google Scholar] [CrossRef] [PubMed]
- De Breuk, A.; Acar, I.E.; Kersten, E.; Schijvenaars, M.M.V.A.P.; Colijn, J.M.; Haer-Wigman, L.; Bakker, B.; de Jong, S.; Meester-Smoor, M.A. Development of a Genotype Assay for Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2021, 128, 1604–1617. [Google Scholar] [CrossRef]
- Chew, E.Y. Revisiting the Question of Genetic Testing for Persons with Age-Related Macular Degeneration. Ophthalmology 2021, 128, 1618–1619. [Google Scholar] [CrossRef]
- Gorin, M.B. Genetic insights into age-related macular degeneration: Controversies addressing risk, causality, and therapeutics. Mol. Asp. Med. 2012, 33, 467–486. [Google Scholar] [CrossRef] [Green Version]
- Dedania, V.S.; Grob, S.; Zhang, K.; Bakri, S.J. Pharmacogenomics of response to anti-VEGF therapy in exudative age-related macular degeneration. Retina 2015, 35, 381–391. [Google Scholar] [CrossRef]
- Hong, N.; Shen, Y.; Yu, C.Y.; Wang, S.Q.; Tong, J.P. Association of the polymorphism Y402H in the CFH gene with response to anti-VEGF treatment in age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2016, 94, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Kozhevnikova, O.S.; Fursova, A.Z.; Derbeneva, A.S.; Nikulich, I.F.; Tarasov, M.S.; Devyatkin, V.A.; Rumyantseva, Y.V.; Telegina, D.V.; Kolosova, N.G. Association between Polymorphisms in CFH, ARMS2, CFI, and C3 Genes and Response to Anti-VEGF Treatment in Neovascular Age-Related Macular Degeneration. Biomedicines 2022, 10, 1658. [Google Scholar] [CrossRef] [PubMed]
- Arslan, J.; Baird, P.N. Changing vision: A review of pharmacogenetic studies for treatment response in age-related macular degeneration patients. Pharmacogenomics 2018, 19, 436–461. [Google Scholar] [CrossRef] [PubMed]
- Abedi, F.; Wickremasinghe, S.; Richardson, A.J.; Islam, A.F.M.; Guymer, R.H.; Baird, P.N. Genetic influences on the outcome of anti-vascular endothelial growth factor treatment in neovascular age-related macular degeneration. Ophthalmology 2013, 120, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Fauser, S.; Lambrou, G.N. Genetic predictive biomarkers of anti-VEGF treatment response in patients with neovascular age-related macular degeneration. Surv. Ophthalmol. 2015, 60, 138–152. [Google Scholar] [CrossRef]
- Oca, A.I.; Pérez-Sala, Á.; Pariente, A.; Ochoa, R.; Velilla, S.; Peláez, R.; Larráyoz, I.M. Predictive Biomarkers of Age-Related Macular Degeneration Response to Anti-VEGF Treatment. J. Pers. Med. 2021, 11, 1329. [Google Scholar] [CrossRef]
- Wickremasinghe, S.S.; **e, J.; Lim, J.; Chauhan, D.S.; Robman, L.; Richardson, A.J.; Hageman, G.; Baird, P.N.; Guymer, R. Variants in the APOE gene are associated with improved outcome after anti-VEGF treatment for neovascular AMD. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4072–4079. [Google Scholar] [CrossRef] [Green Version]
- Yehoshua, Z.; de Amorim Garcia Filho, C.A.; Nunes, R.P.; Gregori, G.; Penha, F.M.; Moshfeghi, A.A.; Zhang, K.; Sadda, S.; Feuer, W.; Rosenfeld, P. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study. Ophthalmology 2014, 121, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Bishop, P.N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 2018, 40, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Holz, F.G.; Sadda, S.R.; Busbee, B.; Chew, E.Y.; Mitchell, P.; Tufail, A.; Brittain, C.; Ferrara, D.; Gray, S.; Honigberg, L.; et al. Efficacy and Safety of Lampalizumab for Geographic Atrophy Due to Age-Related Macular Degeneration: Chroma and Spectri Phase 3 Randomized Clinical Trials. JAMA Ophthalmol. 2018, 136, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Halawa, O.A.; Lin, J.B.; Miller, J.W.; Vavvas, D.G. A Review of Completed and Ongoing Complement Inhibitor Trials for Geographic Atrophy Secondary to Age-Related Macular Degeneration. J. Clin. Med. 2021, 10, 2580. [Google Scholar] [CrossRef]
- Biggs, R.M.; Makou, E.; Lauder, S.; Herbert, A.P.; Barlow, P.N.; Katti, S.K. A novel full-length recombinant human complement factor H (CFH; GEM103) for the treatment of age-related macular degeneration shows similar in vitro functional activity to native CFH. Curr. Eye Res. 2022, 47, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, G.J.; Sahni, J.; Fauser, S.; Geary, R.S.; Schneider, E.; McCaleb, M. Development of IONIS-FB-LRx to Treat Geographic Atrophy Associated with AMD. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4305. [Google Scholar]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Steinle, N.C.; Pearce, I.; Monés, J.; Metlapally, R.; Saroj, N.; Hamdani, M.; Ribeiro, R.; Rosenfeld, P.J.; Lad, E.M. Impact of Baseline Characteristics on Geographic Atrophy Progression in the FILLY Trial Evaluating the Complement C3 Inhibitor Pegcetacoplan. Am. J. Ophthalmol. 2021, 227, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.L.; Pouw, R.B.; Kavanagh, D.; Sun, R.; Ricklin, D. Developments in anti-complement therapy; from disease to clinical trial. Mol. Immunol. 2018, 102, 89–119. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Monés, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology 2021, 128, 576–586. [Google Scholar] [CrossRef]
- Raimondi, R.; Zollet, P.; De Rosa, F.P.; Tsoutsanis, P.; Stravalaci, M.; Paulis, M.; Inforzato, A.; Romano, M.R. Where Are We with RPE Replacement Therapy? A Translational Review from the Ophthalmologist Perspective. Int. J. Mol. Sci. 2022, 23, 682. [Google Scholar] [CrossRef]
- Khanani, A.M.; Thomas, M.J.; Aziz, A.A.; Weng, C.Y.; Danzig, C.J.; Yiu, G.; Kiss, S.; Waheed, N.K.; Kaiser, P.K. Review of gene therapies for age-related macular degeneration. Eye 2022, 36, 303–311. [Google Scholar] [CrossRef]
- Moore, N.A.; Bracha, P.; Hussain, R.M.; Morral, N.; Ciulla, T.A. Gene therapy for age-related macular degeneration. Expert Opin Biol. Ther. 2017, 17, 1235–1244. [Google Scholar] [CrossRef]
- Mullins, R.F.; Schoo, D.P.; Sohn, E.H.; Flamme-Wiese, M.J.; Workamelahu, G.; Johnston, R.M.; Wang, K.; Tucker, B.A.; Stone, E.M. The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol. 2014, 184, 3142–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heier, J.S.; Kherani, S.; Desai, S.; Dugel, P.; Kaushal, S.; Cheng, S.H.; Delacono, C.; Purvis, A.; Richards, S.; Le-Halpere, A.; et al. Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: A phase 1, open-label trial. Lancet 2017, 390, 50–61. [Google Scholar] [CrossRef]
- Reid, C.A.; Nettesheim, E.R.; Connor, T.B.; Lipinski, D.M. Development of an inducible anti-VEGF rAAV gene therapy strategy for the treatment of wet AMD. Sci. Rep. 2018, 8, 11763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- REGENXBIO Announces Additional Positive Interim Phase I/IIa and Long-Term Follow-Up Data of RGX-314 for the Treatment of Wet AMD. Available online: https://www.prnewswire.com/news-releases/regenxbio-announces-additional-positive-interim-phase-iiia-and-long-term-follow-up-data-of-rgx-314-for-the-treatment-of-wet-amd-301228344.html (accessed on 16 February 2021).
- Grishanin, R.; Vuillemenot, B.; Sharma, P.; Keravala, A.; Greengard, J.; Gelfman, C.; Blumenkrantz, M.; Lawrence, M.; Hu, W.; Kiss, S.; et al. Preclinical Evaluation of ADVM-022, a Novel Gene Therapy Approach to Treating Wet Age-Related Macular Degeneration. Mol. Ther. 2019, 27, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Busbee, B.; Boyer, D.S.; Khanani, K.M.; Wykoff, C.C.; Pieramici, D.J.; Regillo, C.; Danzig, C.J.; Joondeph, B.C.; Major, J.; Hoang, C.; et al. Phase 1 Study of Intravitreal Gene Therapy with ADVM-022 for neovascular AMD (OPTIC Trial). Investig. Ophthalmol. Vis. Sci. 2021, 62, 352. [Google Scholar]
- Pecen, P.E.; Kaiser, P.K. Current phase 1/2 research for neovascular age-related macular degeneration. Curr. Opin Ophthalmol. 2015, 26, 188–193. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Lauer, A.K.; Sohn, E.H.; Mir, T.A.; Naylor, S.; Anderton, M.C.; Kelleher, M.; Harrop, R.; Ellis, S.; Mitrophanous, K.A. Vector Gene Transfer of Endostatin/Angiostatin for Macular Degeneration (GEM) Study. Hum. Gene Ther. 2017, 28, 99–111. [Google Scholar] [CrossRef]
- Ren, X.; Li, J.; Xu, X.; Wang, C.; Cheng, Y. IBI302, a promising candidate for AMD treatment, targeting both the VEGF and complement system with high binding affinity in vitro and effective targeting of the ocular tissue in healthy rhesus monkeys. Exp. Eye Res. 2016, 145, 352–358. [Google Scholar] [CrossRef]
- Khandhadia, S.; Cipriani, V.; Yates, J.R.W.; Lotery, A.J. Age-related macular degeneration and the complement system. Immunobiology 2012, 217, 127–146. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Yu, D.; Liu, Q.; Lin, S.; Tian, R.; Li, J.; Luo, Y. Protective Effect of a Bispecific Fc-Fusion Protein on the Barrier of Human Retinal Pigment Epithelial Cells. Ophthalmic Res. 2021, 64, 656–663. [Google Scholar] [CrossRef]
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X.; et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Environmental Factors | No Modifiable | Modifiable |
|---|---|---|
| AGE | SMOKING | |
| EPIGENETICS DNA methylation Chromatine changes Histone acetylation miRNA | NUTRITION | |
| ALCOHOL | ||
| LIGHT EXPOSURE | ||
| HIGH BLOOD PRESSURE |
| Pathway | Molecule | Polymorphism | Evidence |
|---|---|---|---|
| Immune response and complement genes | CFH | rs1061170, rs10922109, rs121913059 | Fritsche et al., 2016 [33], Edwards et al., 2005 [35], Park et al., 2019 [36], Herrsterbeek et al., 2020 [37], Ferreira et al., 2009 [38] |
| C3 | rs2230199, rs1047286 | Maller et al., 2007 [39], Thakkinstian et al., 2010 [40] | |
| CFB | rs641153, rs415667 | Wang et al., 2013 [41], Thakkinstian et al., 2012 [42] | |
| C2 | rs9332739, rs547154 | Wang et al., 2013 [41], Thakkinstian et al., 2012 [42] | |
| C9 | rs62358361, rs34882957 | Fritsche et al., 2016 [33], Seddon et al., 2013 [43], | |
| CFI | rs2285714, rs10033900 | Ven et al., 2013 [44], Wang et al., 2016 [45] | |
| other | PLEKHA/ARMS2/HTRA-1 | rs10490924, rs11200638 | Kanda et al., 2007 [46], Mullins et al., 2019 [47], Zhang et al., 2021 [48], Liu et al., 2020 [49], DeWan et al., 2006 [50], Chen et al., 2009 [51] |
| Oxidative stress genes | MnSOD | Ala-9Val, Ile58Thr | Kimura et al., 2020 [52], Kowalski et al., 2010 [53] |
| HMOX1 HMOX2 | rs2071747, rs2270363 | Synowiec et al., 2012 [54] | |
| Lipid metabolism genes | ApoE | rs429358 | Fritsche et al., 2016 [33] |
| ABCA1 | rs2740488 | Fritsche et al., 2016 [33] | |
| LIPC | rs0468017, rs493258, rs2043085 | Yu et al., 2011 [55], Fritsche et al., 2016 [33] | |
| Neovascularisation genes | VEGF | rs943080 | Zaho et al., 2013 [56], Balikova et al., 2019 [57] |
| Complement | ||||||
|---|---|---|---|---|---|---|
| Antagonist | Therapeutic (Alt. Name) | Pharma Company | Treatment Type | Complement Target | Administration | Clinical Trials |
| GEM 103 | Gemini Therapeutics | full-length recombinant | CFH | Intravitreal injection | NCT04643886 phase II | |
| AMY 101 | Amyndas Pharmaceuticals | peptidic complement inhibitor (mini-FH) | CFH | administrated in healthy male volunteers | ||
| IONIS-FB-LRX, | Ionis Pharma | antisense oligonucleotide encoding CFB | CFB | Subcutaneous | NCT03815825 phase II | |
| Pegcetacoplan (APL-2) | Apellis Pharmaceuticals | cyclic PEG peptide | C3 | Intravitreal injection | NCT03525613 NCT03525600 phase III | |
| LGF 316 + CLG561 | Novartis | IgG1 antibody + anti-properdin antibody | C5 + properdine | Intravitreal injection | NCT02515942 (completed) | |
| ZIMURA (ARC1905) | IVERIC bio | pegylated RNA aptamer | C5 | Intravitreal injection | NCT04435366 phase III | |
| Gene-Therapy | AAVCAGsCD59 (HMR59) | Janssen Research & Development | AAV gene therapy | CD59 | Single intravitreal injection | NCT03585556 NCT03144999 (completed) |
| GT005 | Gyroscope Therapeutics Limited | AAV gene therapy | CFI | Single subretinal injection | NCT03846193 phase I NCT04566445 NCT04437368 phase II |
| Anti-Angiogenesis | |||||
|---|---|---|---|---|---|
| Gene Therapy | Therapeutic (Alt. Name) | Pharms Company | Gene Expression | Administration | Clinical Trials |
| AAV2-sFLT01 | Genzyme, a Sanofi Company (Modena, Italy) | sFLT01 | Single intravitreal injection | NCT01024998 phase I | |
| RGX-314 | Regenxbio, Inc. Rockville, MD, USA | monoclonal antibody fragment similar to ranizumab | Subretinal via transvitreal injection | NCT04514653, NCT04832724, NCT03999801, NCT05210803, NCT04704921 | |
| ADVM-022 | Adverum Biotechnologies (Redwood City, CA, USA) | coding sequence for aflibercept | Single intravitreal injection | NCT03748784 phase I | |
| 4D-150 | 4D Molecular Therapeutics (Emeryville, CA, USA) | miRNA targeting VEGF-C and sequences encoding aflibercept | Single intravitreal injection | NCT05197270 phase I | |
| RetinoStat (OXB-201) | Oxford BioMedica (Oxford, UK) | endostatin and angiostatin | Intravitreal injection | NCT01678872 phase I | |
| IBI302 | Innovent Biologics Co. Ltd. (Suzhou, China) | decoy receptor fusion protein | Intravitreal injection | NCT04820452 phase II | |
| BD311 | Shanghai BDgene Co., Ltd. (Shanghai, China) | expressing VEGFA antibody | Intravitreal injection | NCT05099094 phase I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stradiotto, E.; Allegrini, D.; Fossati, G.; Raimondi, R.; Sorrentino, T.; Tripepi, D.; Barone, G.; Inforzato, A.; Romano, M.R. Genetic Aspects of Age-Related Macular Degeneration and Their Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 13280. https://doi.org/10.3390/ijms232113280
Stradiotto E, Allegrini D, Fossati G, Raimondi R, Sorrentino T, Tripepi D, Barone G, Inforzato A, Romano MR. Genetic Aspects of Age-Related Macular Degeneration and Their Therapeutic Potential. International Journal of Molecular Sciences. 2022; 23(21):13280. https://doi.org/10.3390/ijms232113280
Chicago/Turabian StyleStradiotto, Elisa, Davide Allegrini, Giovanni Fossati, Raffaele Raimondi, Tania Sorrentino, Domenico Tripepi, Gianmaria Barone, Antonio Inforzato, and Mario R. Romano. 2022. "Genetic Aspects of Age-Related Macular Degeneration and Their Therapeutic Potential" International Journal of Molecular Sciences 23, no. 21: 13280. https://doi.org/10.3390/ijms232113280