1. Introduction
3'5'-cyclic nucleotide phosphodiesterases (PDEs) catalyze the hydrolysis of the secondary messengers adenosine (cAMP) and guanosine monophosphate (cGMP). Thus PDEs terminate intracellular signaling cascades which are related to various physiological processes such as immune responses, cardiac and smooth muscle contraction, visual response, ion channel conductance, apoptosis, and growth control [
1,
2]. Currently, 11 different families of PDEs are identified in mammals based on their primary amino acid and nucleotide sequences, regulatory properties, and substrate specificity [
3,
4,
5] which hold great potential as drug targets for treatment of specific disease states [
6].
The development of inhibitors of PDE10A, the only known member of the PDE10 family, for neurological and psychiatric disorders makes this enzyme an interesting target for molecular imaging approaches. Although studies on expression and pharmacological intervention suggest a relation between PDE10A and striatal hypofunction, the particular pathways which link PDE10A function and neurotransmission are not completely understood. In the brain, PDE10A shows the most unique distinctive expression pattern of all PDEs with particularly high mRNA levels in caudate nucleus and nucleus accumbens, and much lower levels in other CNS regions [
7,
8,
9]. The graduation in transcript expression closely resembles the distribution of the enzyme assessed in detail by immunohistochemistry in different mammalian species [
7,
9,
10]. Within the striatal area, PDE10A was shown to be highly expressed by the GABAergic medium spiny projection neurons which represent ~90% of striatal neurons [
10]. The primary association of PDE10A with the post-synaptic membrane, indicated by subcellular fractionation, appears to link PDE10A activity with the regulation of excitability of medium spiny neurons [
11]. Outside the brain, the level of PDE10A transcripts correlates with PDE10A immunoreactivity as well. Although high expression in develo** spermatocytes, thyroid and pituitary gland as well as striated and cardiac muscle was detected [
6,
7,
8,
10], the physiological function of PDE10A in these peripheral organs is not known.
In accordance with the high expression of PDE10A in striatal regions, the systemic administration of papaverine, a potent PDE10A inhibitor, induced a rapid increase in striatal levels of cGMP and cAMP in mice [
12] and demonstrated a broad-spectrum efficacy in a range of antipsychotic models [
12,
13]. Genetic deletion of PDE10A confirmed not only the involvement of this enzyme in the papaverine-mediated effects but has been reported to produce behavioral responses consistent with increased striatal output [
14,
15]. Furthermore, alterations in the levels of PDE10A transcripts have been related to synaptic plasticity in the hippocampus following longterm potentiation [
16] and in animal models of neurodegenerative disorders [
17].
These data form the basis for the hypothesis that PDE10A inhibitors will have potential for the treatment of neuropsychiatric disorders in humans that are correlated with striatal hypofunction such as schizophrenia, obsessive-compulsive disorders, Parkinson’s disease, and Huntington’s disease, and make PDE10A an interesting target for diagnostic and therapeutic monitoring by non-invasive imaging techniques such as positron emission tomography (PET). Therefore, numerous compounds which have been investigated concerning PDE10A inhibitory potency and behavioral impact [
18,
19,
20,
21,
22] provide the current basis for the development of PDE10A targeting PET radiotracers.
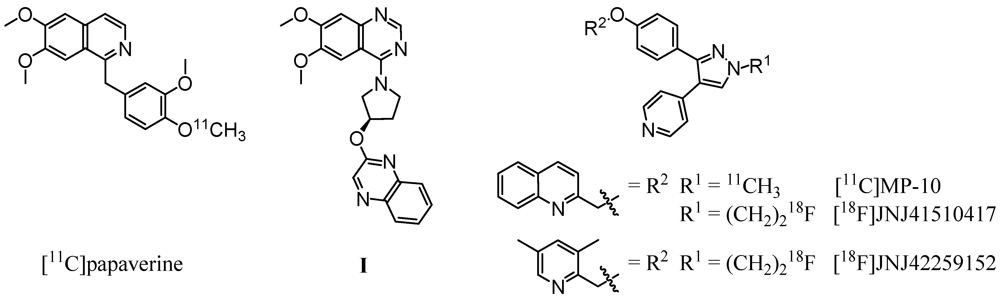
Based on the PDE10A inhibitor papaverine (IC
50,PDE10A = 36 nM [
12]), Tu
et al. successfully synthesized a carbon-11 labeled derivative (
Figure 1, left) by methylation of the corresponding 4-phenolate with [
11C]methyl iodide [
23]. Evaluation
in vivo revealed an initially higher uptake in striatum in comparison to other brain regions. However, the very rapid clearance of the radioligand from the target region indicated [
11C]papaverine as not suitable for imaging of PDE10A. A major progress was made by the same group [
24] and Plisson
et al. [
25] with the development of a carbon-11 labeled analogue of the highly potent and selective PDE10A inhibitor MP-10 (IC
50,PDE10A = 1.26 nM [
13], 0.37 nM [
26]). Methylation of the
N-desmethyl pyrazole precursor with [
11C]methyl iodide led to [
11C]MP-10 (
Figure 1, right), showing a maximum striatum-to-cerebellum ratio of 6.55 in rats and 1.5 to 2 in rhesus monkey at 30 min post-injection (p.i.) [
24]. PET studies of [
11C]MP-10 in pig and baboon revealed a specific, discrete, and reversible uptake in striatum with a peak at 1–2 min p.i. in porcine and at 40–60 min p.i. in nonhuman primate brain [
25].
Figure 1.
Radioligands investigated for PDE10A imaging and compound I, used as lead structure in this study.
Figure 1.
Radioligands investigated for PDE10A imaging and compound I, used as lead structure in this study.
Enabling high-resolution and long-term kinetic PET studies,
18F-labeled MP-10 derivatives were developed as well. (
Figure 1, right). The
N-[
18F]fluoroethyl pyrazole derivative [
18F]JNJ41510417 was synthesized by Celen
et al. [
27,
28] using direct nucleophilic substitution of the corresponding
O-mesylated precursor with n.c.a. [
18F]fluoride (JNJ41510417 IC
50,PDE10A = 0.5 nM [
28], pIC
50,PDE10A = 9.3 [
29]). This radioligand showed reversible and PDE10A specific striatal binding in biodistribution as well as in dynamic small-animal PET studies with a maximum striatum-to-cerebellum ratio of 4.60 at 30 min p.i. [
29]. Target specific binding was confirmed by imaging in PDE10A knockout mice. Nevertheless, high plasma protein binding of [
18F]JNJ41510417, probably due to its high lipophilicity (JNJ41510417 c log
P = 4.19 [
29]), and detrimental slow kinetics have been observed [
28]. This result prompted the same group to synthesize the smaller and less lipophilic dimethylpyridine analogue [
18F]JNJ42259152 (JNJ42259152 c log
P = 3.66, pIC
50,PDE10A = 8.8) [
29,
30] via alkylation of the
N-desmethyl pyrazole precursor with [
18F]fluoroethyl bromide. Biodistribution studies in rats demonstrated a striatum-to-cerebellum ratio of 5.38 at 30 min p.i. Dynamic small-animal PET imaging in rat and monkey showed highly intensive, reversible and PDE10A selective uptake of the radioligand in striatum, with low background activity in cortical regions and cerebellum [
30]. In consequence, a first human study is planned for PDE10A imaging with [
18F]JNJ42259152.
However, all MP-10 derived radioligands developed so far are adversely affected by metabolism [
24,
25,
28,
29,
30]. Fast metabolism was observed in plasma of rats [
28], baboon [
24,
25] and rhesus monkey [
30]. The corresponding phenolic radioactive metabolite passed the blood-brain-barrier in rat and accumulated in the brain up to 15.9% [
24]. In cerebellum up to 25.8% [
28] and 53% [
30] of metabolites were found at 60 min p.i.
As a structurally alternative approach and to possibly overcome the metabolic stability problem of the described PDE10A ligands, we intended to develop radioligands for PET imaging of PDE10A based on a 6,7-dimethoxyquinazoline (
I,
Figure 1, center). Published by a Pfizer research group in 2007, this potent papaverine-related ligand inhibited PDE10A with a
Ki,PDE10A of 4 nM [
31]. In this study we report on the development and investigation of a fluorine-18-labeled analogue. In a series of
I-based compounds, the reference compound (
R)-7-(2-fluoroethoxy)-6-methoxy-4-(3-(quinoxalin-2-yloxy)pyrrolidin-1-yl)quinazoline as well as two potential labeling precursors were synthesized in multi-step syntheses, and their PDE inhibitory potency was investigated [
32] (data will be published in detail elsewhere). Since the 7-fluoroethoxyquinazoline derivative showed acceptable inhibitory potency with a
Ki,PDE10A of 53 nM and occurred as radiochemically most accessible, this compound was chosen for radiolabeling with fluorine-18 and subsequent evaluation
in vitro and
in vivo.
3. Experimental Section
3.1. General
No-carrier-added [18F]fluoride (half-life: 109.8 min) was produced via the [18O(p, n)18F] nuclear reaction by irradiation of a [18O]water target (>97% enriched, 2 mL) on a PETtrace cyclotron (16.5 MeV proton beam), GE Healthcare. The final radiolabeled product was purified by semi-preparative HPLC on a TRACERlabTM FX F-N synthesizer (GE Healthcare, Waukesha, WI, USA) containing a S1021 pump (SYKAM Chromatographie Vertriebs GmbH, Fürstenfeldbruck, Germany); WellChrom K-2001 UV detector (KNAUER GmbH, Berlin, Germany); NaI(Tl)-counter; automated data acquisition, NINA, Nuclear Interface) and by solid phase extraction on SepPak®Plus cartridges (Waters Corporation, Milford, MA, USA). Analyses by TLC were performed on POLYGRAM® SIL G/UV254 and POLYGRAM® ALOX N/UV254 plates, 40 × 80 mm (MACHEREY-NAGEL GmbH & Co. KG, Düren, Germany). Analytical HPLC was carried out on a computer assisted LC-2000Plus system from JASCO International Co., Ltd. (including LC-NetII/ADC Netbox, DG-2080-54 4-Line Degasser, LG-2080-04S quaternary gradient unit, PU-2080Plus HPLC Pump, AS-2055Plus Sampler, UV-2070Plus UV/Vis Detector), coupled with a radioactivity-HPLC-flow-monitor (Gabi Star, Raytest GmbH, Straubenhardt, Germany).
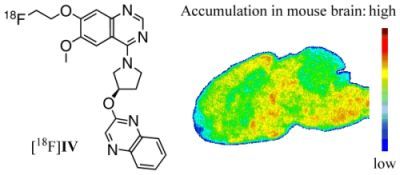
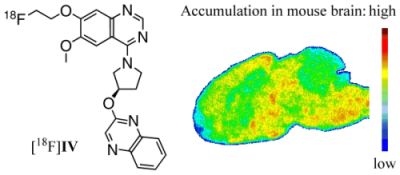
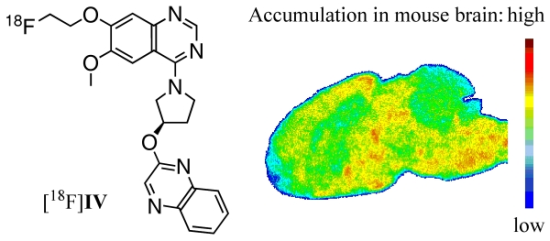
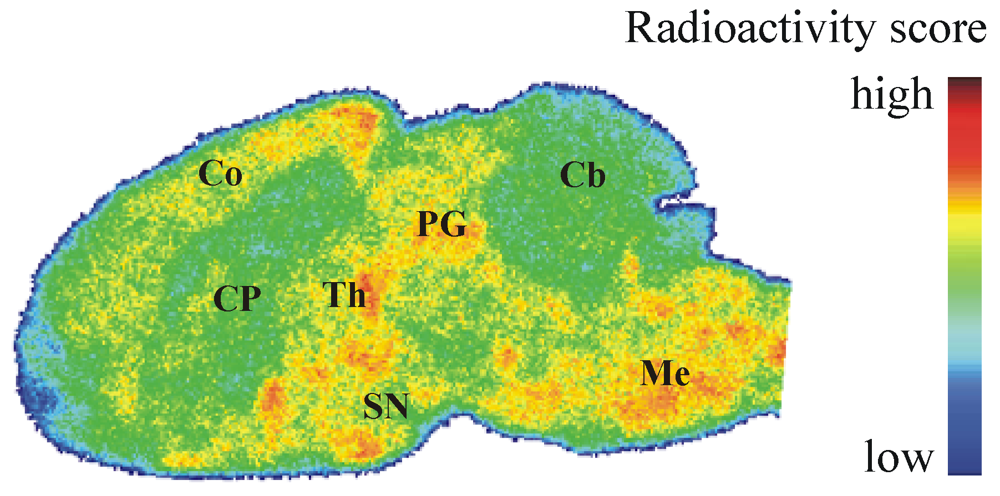
Figure 5.
Representative color-coded autoradiographic image of a sagittal mouse brain slice. Distribution of [18F]IV in mice brain ex vivo, obtained at 30 min p.i. of 100 MBq. Co: cortex; CP: caudate putamen; PG: periaqueductal gray; Th: thalamus; SN: substantia nigra; Cb: cerebellum; Me: Medulla.
Figure 5.
Representative color-coded autoradiographic image of a sagittal mouse brain slice. Distribution of [18F]IV in mice brain ex vivo, obtained at 30 min p.i. of 100 MBq. Co: cortex; CP: caudate putamen; PG: periaqueductal gray; Th: thalamus; SN: substantia nigra; Cb: cerebellum; Me: Medulla.
Processed TLC plates and organ sections were exposed to 18F-sensitive storage phosphor screens (BAS-TR2025, FujiFilm Co., Tokyo, Japan), and image plates were analyzed using a bioimaging analyzer system (BAS-1800 II, BASReader 2.26 Beta and AIDA 2.31 Image Analyzer software; Raytest GmbH, Straubenhardt, Germany, and FujiFilm Co., Tokyo, Japan).
In vivo studies were carried out in 3-months-old female CD-1 mice (20–25 g). Animals were obtained from the Medizinisch-Experimentelles Zentrum, Universität Leipzig, maintained on a 12 h light-dark cycle, and habituated for at least two days before experiments. All procedures involving animals were approved by the respective State Animal Care and Use Committee and conducted in accordance with the German Law for the Protection of Animals.
3.2. Radiochemistry
3.2.1. Two-Step Radiosynthesis of (R)-7-(2-[18F]Fluoroethoxy)-6-methoxy-4-(3-(quinoxalin-2-yloxy)pyrrolidin-1-yl)quinazoline ([18F]IV)
A representative procedure is given as follows. For [
18F]fluoride activation, colorless aqueous n.c.a. [
18F]fluoride solution (1–2 mL) was added to 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo-[8.8.8]hexacosane (Kryptofix
®, K2.2.2, 11.20 mg, 0.03 mmol), 0.15 M aqueous K
2CO
3 solution (89 μL, 0.013 mmol) and acetonitrile (1 mL) in a 10 mL conical vessel, equipped with magnetic stirrer, septum, as well as vacuum and inert gas access. This mixture was azeotropically dried according to previously described protocols [
41,
42], resulting in the K[
18F]F-K2.2.2-carbonate complex, dissolved in anhydrous acetonitrile (1 mL). With 500 μL of the light yellow solution, nucleophilic substitution was performed in a 5 mL conical vessel under inert gas by reacting ethane-1,2-diyl bis(4-methylbenzenesulfonate)
II (2.0 mg, 0.005 mmol,
Scheme 1) under stirring at 80 °C, using a heating block. To monitor the fluorination step, 15 μL aliquots were taken, diluted and analyzed (radio-HPLC, isocratic elution with 45% MeCN/H
2O and 15 mM AcOH/12.5 mM TEA, flow rate 1 mL/min, λ = 245 nm; Kromasil 100 C18, 5 μm, 250 × 4.6 mm, JASCO International Co., Ltd., t
R,[18F]III = 15 min; radio-TLC, SiO
2 with petroleum ether/EtOAc/NH
3aq. 25%, 5/5/0.25, v/v/v, R
f,[18F]III = 0.72). Thus, a labeling efficiency of 75% was observed after 5 min of reaction.
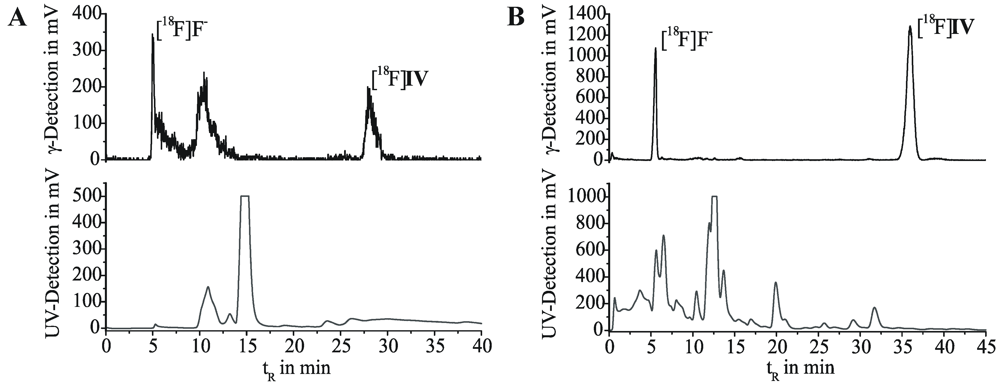
In a 2 mL reaction vessel, equipped with magnetic stirrer, septum and inert gas access, (R)-7-hydroxy-6-methoxy-4-[3-(quinoxalin-2-yloxy)pyrrolidine-1-yl]quinazoline (V, 4.0 mg, 0.01 mmol) in acetonitrile (1 mL) were stirred with Kryptofix® (1.90 mg, 0.01 mmol) and K2CO3 (0.35 mg, 0.005 mmol) at 60 °C, using a heating block. After 2 h, the acetonitrile was carefully evaporated to dryness under inert gas and the mixture dissolved in anhydrous acetonitrile (1 mL). At room temperature, the yellow phenolate-containing solution (500 μL, 0.005 mmol) was added to a solution of [18F]III (500 μL) in a conical 6 mL vessel and stirred under inert gas at 80 °C, using a heating block. For reaction control, aliquots of 15 µL were taken, diluted and analyzed on radio-TLC (SiO2 with MTBE/MeOH/NH3aq. 25%, 10/1/0.25, v/v/v, Rf,[18F]IV = 0.45), as well as radio-HPLC (isocratic elution with 45% MeCN/H2O and 15 mM AcOH/12.5 mM TEA, flow rate 1 mL/min, λ = 245 nm; Kromasil 100 C18, 5 μm, 250 × 4.6 mm, JASCO International Co., Ltd., tR,[18F]IV = 12 min). After 15 min reaction time, labeling efficiencies of ~38% were observed.
The crude, ochre-coloured reaction mixture was cooled to room temperature, diluted with water (50 mL) and passed through a neutral Al2O3 cartridge. The crude product [18F]IV was eluted with acetonitrile (1.5 mL), diluted to 4 mL with water and subjected to semi-preparative radio-HPLC (isocratic elution with 55% MeCN/H2O, 20 mM NH4OAc; λ = 254 nm; flow rate 2 mL/min on a RP-column with precolumn, 50 × 10 mm and 150 × 10 mm, Multospher 120 RP-18 AQ, CS-Chromatographie Service GmbH, Langerwehe, Germany; tR,[18F]IV = 28 min). Fractions containing [18F]IV were collected and analyzed on radio-TLC as well as radio-HPLC. After 3.5 h for the entire process, the final product was obtained with a decay corrected radiochemical yield of 29% (based on cyclotron produced [18F]F−) and a radiochemical purity of 99%, maintaining unspecified chemical impurities.
3.2.2. One-Step Radiosynthesis of [18F]IV
A representative procedure is given as follows. The K[
18F]F-K2.2.2-carbonate complex was prepared as described in
Section 3.2.1. and dissolved in anhydrous acetonitrile (500 μL). Under inert gas, the light yellow solution (250 μL) was added to (
R)-2-(6-methoxy-4-(3-(quinoxalin-2-yloxy)pyrrolidin-1-yl)quinazolin-7-yloxy)ethyl 4-methylbenzenesulfonate (
VI, 2.1 mg, 0.004 mmol,
Scheme 1), dissolved in acetonitrile (250 μL), and stirred at 80 °C, using a heating block. Reaction monitoring was done by taking 15 μL aliquots which were diluted and analyzed on radio-TLC (SiO
2 with MTBE/MeOH/NH
3aq. 25%, 9/1/0.25, v/v/v, R
f,[18F]IV = 0.72; Al
2O
3 with cyclohexane/EtOAc, 1/9, v/v, R
f,[18F]IV = 0.63), as well as radio-HPLC (gradient elution with 10 to 90% MeCN/H
2O and 20 mM NH
4OAc over 55 min, flow rate 1 mL/min, λ = 245 nm; Reprosil Gold C18, 5 μ, 250 × 4.6 mm, Maisch HPLC GmbH, Ammerbuch-Entringen, Germany, t
R,[18F]IV = 31 min). Thus, 10 min of reaction led to a labeling efficiency of 60%.
At room temperature, the ochre-coloured reaction mixture was diluted with water (50 mL) and passed through a SiO
2 or C-18 cartridge, and crude [
18F]
IV was eluted with acetonitrile (1.25 mL). After dilution to 4 mL with water, the cloudy solution was manually injected to a semi-preparative radio-HPLC. During isocratic elution (50% MeCN/H
2O, 20 mM NH
4OAc; λ = 254 nm; flow rate 1.5 mL/min on a RP-column with precolumn, 50 × 10 mm and 150 × 10 mm, Reprosil-Pur C18-AQ, 7 μ, Maisch HPLC GmbH, Ammerbuch-Entringen, Germany; t
R,[18F]IV = 32 min), fractions containing [
18F]
IV were collected and analyzed with radio-TLC, as well as radio-HPLC (see above). The combined fractions were diluted with water (40 mL) and passed through a C-18 cartridge. After washing with water (2 mL), [
18F]
IV was eluted with ethanol (1.25 mL) and the solvent was carefully evaporated under argon. The desired radiotracer was then dissolved in a solution of 5% ethanol in a 0.9% sodium chloride isotonic solution. Within 3.3 h for the entire process, [
18F]
IV was obtained in a decay corrected radiochemical yield of 40% (based on cyclotron produced [
18F]F
−), a chemical and radiochemical purity of ≥99%, and specific activity of 269 GBq/μmol (determination via analytical HPLC with UV/mass calibration). The final product [
18F]
IV was shown by radio-TLC (see above) and analytical radio-HPLC (gradient elution with 10% to 90% MeCN/H
2O and 20 mM NH
4OAc over 80 min, flow rate 1 mL/min, λ = 245 nm; Reprosil Gold C18, 5 μ, 250 × 4.6 mm, Maisch HPLC GmbH, Ammerbuch-Entringen, Germany, t
R,[18F]IV = 39 min) to be identical to the authentic non-radioactive material
IV [
32] and to be free of significant chemical and radiochemical impurities.
3.3. Determination of Lipophilicity of [18/19F]IV
3.3.1. Determination of Log D7.2–7.4 Values by Shake-Flask Method
Log D7.2–7.4 values of [18F]IV were determined using a conventional shake-flask method (multiple distribution in triplicates). Measurements of distribution coefficients were performed in 3 series at 21 °C between n-octanol and three aqueous buffer solutions, physiological phosphate-saline (0.1 M Na3PO4, 0.15 M NaCl, pH 7.2), phosphate buffer (KH2PO4/Na2HPO4•2H2O, 50 mM, pH 7.2) or TRIS-HCl buffer (50 mM, pH 7.4 at 21 °C). Solutions of [18F]IV (2–5 MBq) in MeCN, MeOH (100 μL) or in 5% ethanol/0.9% NaCl (10 μL) were added to a 15 mL polypropylene tube (n = 3 per buffer system) containing a two-layer system of a selected aqueous solution (3 mL) and n-octanol (3 mL). Both phases were presaturated with the corresponding aqueous solution or n-octanol, respectively. The tubes were shaken for 30 min (200 rpm, HS250 basic, IKA® Labortechnik GmbH & Co. KG, Staufen, Germany) and then centrifuged at 6000 rpm (21 °C, 5 min; EBA 12R, Andreas Hettich GmbH & Co. KG, Tuttlingen, Germany). Aliquots of 1 mL of the n-octanol phase and 1 mL of the aqueous phase were transferred into separate sample tubes with care. Radioactivity of these samples was quantified using a calibrated γ-counter (Wallac WIZARD, Perkin Elmer, Waltham, MA, USA). The procedure mentioned above was repeated for each serial 3–4 times using 1 mL of the n-octanol phase collected after each extraction step. Distribution coefficients and log D7.2–7.4 values of [18F]IV were calculated with MicrocalTM Origin (6.0, Microcal Software, Inc., Northhampton, MA, USA).
3.3.2. Determination of log D7.0 Values by RP-HPLC Retention
Referring to an OECD guideline [
36] and using a series of reference compounds (nitrobenzene,
p-nitrophenol, benzene, toluene, chlorobenzene, benzophenone, naphthalene, 1,4-dibromobenzene, biphenyl, diphenylether, 1,4-diiodobenzene) with known log
POW values comprised between 1.89 and 4.64, retention times were determined on two reversed phase columns (Multospher 120 RP-18 AQ, 250 mm × 4.6 mm, 5 μm, CS-Chromatographie Service GmbH, Langerwehe, Germany; SupelcosilTM ABZ + Plus, 250 × 4 mm, 5 μm, Supelco, Bellefonte, PA, USA). On an 1100 HPLC ChemStation (Agilent Technologies, Santa Clara, CA, USA) using a gradient (5% to 80% MeCN/H
2O and 20 mM NH
4OAc over 65 min, pH = 7.0, flow rate 1 mL/min, λ = 254 nm), reference compound
IV (t
R,IV@Multospher = 43.7, t
R,IV@Supelcosil = 27.9 min) as well as standard compounds have been eluted with n ≥ 3 on each column. Dead times t
0 were determined by retention of thiourea. Corresponding capacity factors k for all compounds were calculated and plotted as log k
versus log
P of the standard compounds, affording calibration curves (Microcal
TM Origin 6.0, Microcal Software, Inc., Northhampton, MA, USA). The regression equations were then used to calculate the log
D7.0 values of
IV via its corresponding log k (values are given in
Table 1).
3.4. In Vitro Characterization
3.4.1. PDE10A Affinity
Crude membrane homogenates isolated from PDE10A-transfected SF21 cells were obtained from Biocrea GmbH, Radebeul, Germany [
43]. For competitive radioligand displacement experiments (n = 2), the membrane preparations were diluted with incubation buffer (50 mM Tris-HCl, pH 7.4 at room temperature, 5 mM MgCl
2) and re-homogenized by passing through a 27-gauge needle. The protein (100 µg) was incubated with 0.05–0.1 nM of [
18F]
IV and different concentrations of
IV (10 pM–1 µM), diluted from a 10 mM stock in DMSO, at a final volume of 1 mL. Nonspecific binding was determined in the presence of 10 µM of
I, and control incubation was carried out accordingly using crude membrane preparations obtained from non-transfected SF21 cells.
Incubation at room temperature was terminated after 120 min by rapid filtration (Brandel 48-channel harvester; Biomedical Research and Development Lab. Inc., Gaithersburg, MD, USA) on Whatman GF/B glass-fiber filters, pre-soaked for 90 min in 0.3% polyethylenimine (Sigma, Deisenhofen, Germany), followed by washing four times with 4 mL of ice-cold wash buffer (50 mM Tris-HCl, pH 7.4 at 4 °C). Filter-bound radioactivity was quantified using a calibrated γ-counter (Wallac WIZARD, Perkin Elmer, Waltham, MA, USA).
3.4.2. Data Analysis
Each experiment was carried out in triplicate. The binding parameter
IC50 was estimated using iterative non-linear curve fitting.
KD of [
18F]
IV was estimated directly from homologous competition curve with
KD = IC
50-[radioligand] as well as from the respective saturation isotherm by nonlinear regression analysis [
37].
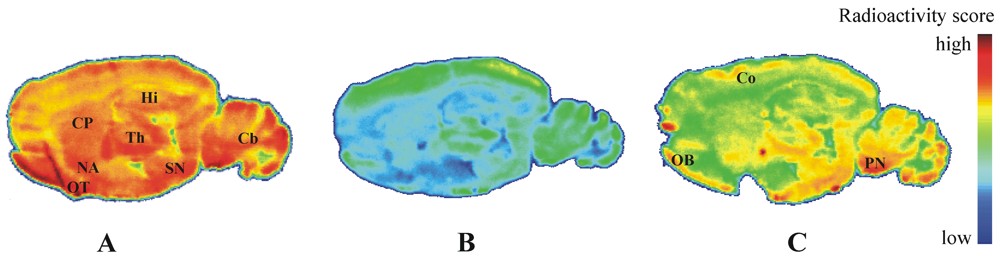
3.4.3. In Vitro Autoradiographic Study
Frozen sagittal brain sections (12 μm) obtained from female SPRD rats (8–10 weeks) were thawed, dried in a stream of cold air, preincubated for 20 min with 50 mM TRIS-HCl, pH 7.4, at room temperature, and dried again. For determination of total binding, sections were incubated with a 25 nM solution of [
18F]
IV in 50 mM TRIS-HCl, pH 7.4, at room temperature for 60 min. Nonspecific binding was determined in the presence of 100, 10, and 1 µM of
IV or MP-10 [
13,
26] (MP-10 obtained as a gift from Bjarke Ebert, H. Lundbeck A/S, Valby Denmark). After incubation, sections were washed twice for 2 min with 50 mM TRIS-HCl, pH 7.4 at 4 °C, dipped briefly in ice-cold deionized water (5 s), dried in a stream of cold air, and exposed for 17 h to
18F-sensitive storage phosphor screens, which were analyzed using the BAS-1800 II system (see above).

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


