1. Introduction
The majority of infectious diseases that affect humans are caused by bacteria [
1]. Despite recent advances in antimicrobial medicine, bacterial infections are still a major cause of mortality worldwide. On the basis of their infective lifestyle, pathogenic bacteria are classified into three major groups: extracellular, facultative intracellular, and obligatory intracellular. Bacteria that live outside the host are known as extracellular or free-living bacteria, while those that have both intracellular and extracellular phases (capable of replicating inside and outside a host cell) are known as facultative intracellular bacteria [
1]. Obligatory intracellular pathogenic bacteria are distinct from the preceding two bacteria in that they have restricted characteristics that necessitate a host for reproduction [
1]. A better grasp of the complicated interplay between hosts and pathogens will help create new preventive and therapeutic treatments that can be applied to resolve infection. Accurate classifications of infectious agents have been developed that take the infective lifestyle of various pathogens and their specificities of hosts into consideration [
2]. For the purpose of organizing fruitful future research, this knowledge is crucial.
Many bacteria, such as
Escherichia coli (
E. coli),
Haemophilus influenzae (
H. influenzae),
Mycoplasma spp.,
Pseudomonas aeruginosa (
P. aeruginosa),
Streptococcus pyogenes (
S. pyogenes),
Vibrio cholerae (
V. cholerae), etc., are extracellular, possessing the ability to reproduce in extracellular space. Extracellular bacteria are normally eliminated from the body by macrophages (M), neutrophils, and dendritic cells (DCs) through phagocytosis. However, those that survive may potentially be hazardous, as they release toxins and trigger excessive inflammatory responses, damaging and destroying host cells and tissues. Comparatively, intracellular bacteria have developed adaptations that allow them to avoid lysosome-mediated degradation in host cells, allowing them to replicate. The body has evolved distinct immune responses, such as T cell activation, particularly CD8+ T cells, to resolve such infections. Facultative intracellular bacteria can survive both inside and outside of host cells. They include
Legionella pneumophila (
L. pneumophila),
Salmonella spp.,
Mycobacterium tuberculosis (
M. tuberculosis),
Neisseria meningitidis (
N. meningitidis),
Shigella dysenteriae (
S. dysenteriae),
Francisella tularens (
F. tularens),
Bordetella pertussis (
B. pertussis), and
Bacillus anthracis (
B. anthracis). On the other hand, obligate intracellular bacteria are wholly dependent on the host cell machinery for their existence and division.
Chlamydia spp.,
Anaplasma spp.,
Ehrlichia spp.,
Coxiella spp., and all rickettsial and Orientia species are examples of obligate intracellular bacteria. These bacteria cause fatal diseases in humans. The incubation period for different bacteria varies. Some bacteria such as
S.
flexneri,
S. typhimurium, and
S. enterica have a short 12 h incubation, while others such as
M. leprae have longer incubation periods that may last 4–5 years (
Table 1). It is important to note that alternative pathogen classification methods, based on criteria such as molecular ty**, 16S rRNA, and phylogenetics also exist in the literature. Conventional classifications are primarily based on the infective lifestyles of pathogens in the host; however, there are many inconsistencies in this approach. For example, some extracellular bacteria are known to also have an intracellular in vivo phase. The ability of such pathogens to grow in artificial cell-free media has often been exaggerated [
2]. In this context, the term “facultative” has not been adequately explained, as the ability of a pathogen to grow intracellularly during infectious processes has been termed facultative, while in reality, the pathogen characteristics are that of obligate bacteria, and therefore essential. In contrast the term “obligate” is based on the inability to survive in artificial conditions [
2]. This particular inconsistency in bacterial classification has since been resolved by researchers who examined the role of peptidoglycan recognition proteins (PGRPs) in
Drosophila [
3,
4]. PGRPs are pattern recognition molecules that discern unique peptidoglycans in bacterial cell walls. PGRP-LE and PGRP-LC in particular, are involved in the identification of extracellular and intracellular bacteria that have diaminopimelic acid (DAP) type peptidoglycans. They trigger a variety of innate immune responses, including the generation of antimicrobial peptides, melanization, and autophagy.
Vaccines and antibiotics have significantly improved global health. Vaccines prevent fatal infections, while antibiotics have been pivotal in the treatment of life-threatening and often fatal infectious diseases caused by intracellular bacteria. The development of vaccines against intracellular microorganisms faces numerous difficulties. The genetic diversity found in bacterial strains poses one of the most profound obstacles. The emergence of antibiotic resistance is another challenge that further increases the genetic diversity of bacteria. Furthermore, as the capsular polysaccharide of bacteria has weak immunogenicity and high similarity to fetal neural tissues, the development of vaccines against bacteria such as N. meningitidis (serogroup B vaccine) is more challenging. The rise in intracellular bacteria that are resistant to inactivated whole cell vaccines also presents an additional layer of difficulty in vaccine development. Therefore, to overcome these difficulties and challenges, novel approaches are required for the development of anti-intracellular bacteria vaccines.
2. Traditional Vaccine Development
The fundamental concept of vaccine development postulated by Louis Pasteur towards the end of the 19th century serves as the cornerstone of classical vaccination (isolation, inactivation, and injection of the causal agent). Throughout the 20th century, this paradigm drove the development of vaccines [
5]. The conventional method has helped develop vaccines that have led to the eradication of diseases such as smallpox, pertussis, measles, mumps, rubella, and Haemophilus influenzae B, while generally increasing life expectancy. Based on these empirical principles, three conventional methods are employed involving the application of inactivated microorganisms, live attenuated agents, and pathogen subunits for vaccine development against different infections (
Table 2) [
6]. Inactivated vaccines contain severe disease-causing bacteria that have lost their capacity to infect or multiply inside or outside of a host. Such vaccines can be generated using physical, chemical, or radiation-based techniques that do not compromise microbe antigenicity. As inactivated microorganisms do not survive in the extracellular environment or within vaccinated individuals, they are generally considered safe. However, compared to other vaccine types, their application may offer reduced or short-lasting protection. Examples of such vaccines include
Brucella melitensis Rev1 (brucellosis prevention in sheep and goats), Dukoral (cholera vaccine), Shanchol (cholera vaccine), Q-Vax (
C. burnetii), and many others (
Table 2). Live attenuated vaccines are comparatively more efficient against intracellular bacteria as they contain live bacteria or viruses that have been “weakened” (attenuated) to elicit a protective immune response without inflicting disease in healthy individuals. This “weakening” of the pathogen is accomplished by inducing genetic change. Genetic changes that reduce virulence may be induced in the laboratory by repeatedly culturing the pathogen (serial passage), or by introducing the pathogen to a non-specific “foreign” host in which genetic variability and mutation cause the pathogen to lose its virulence to the natural host. Most modern vaccines contain live, attenuated pathogens; examples include the
M. bovis strain
Bacillus Calmette–Guerin (BCG) vaccine, vaccines against
S. enterica spp., the
B. anthracis vaccine (BioThrax), and the
V. cholerae vaccine (Vaxchora) (
Table 2). Subunit vaccines contain purified antigenic parts of a pathogen (instead of the whole organism) that elicit a host immune response. Purified antigens can be toxoids, subcellular fragments, or surface molecules, and they can be conveyed by a variety of carriers. Compared to live attenuated vaccines, the immune responses elicited by subunit vaccines are not as potent or long lasting. Repeated doses are often required, followed by booster doses in the following year. Subunit vaccinations frequently have adjuvants added. These are elements that support and prolong the immunological response to the vaccine. The majority of newly developed leishmaniasis vaccines target different antigens using protein subunits. Tularemia subunit vaccines have also been created in which the
F. tularensis surface protein Tul4 and the heat shock protein DNAK were combined and delivered to mice intranasally together with the adjuvant GPI to demonstrate long-term pathogen-specific immunity [
7]. Other conventional methods used for vaccine development along with their respective status are summarized in
Table 2.
Conventional vaccine development strategies are often associated with various limitations such as short duration of effectiveness, numerous safety concerns, and high production costs. Inactivated and attenuated vaccine development strategies often face delays owing to the difficulties associated with the culturing of some bacterial strains. Some vaccines may not be adequately attenuated and thus may still elicit detrimental immune responses. With respect to subunit vaccines, the antigen purification process is often costly and technically challenging. While the effectiveness of traditional vaccines may be disease dependent, new diseases are also emerging due to mutations, gene exchange, interspecies transfer, and novel environmental exposures. Therefore, more reliable and novel methods are required to counter these challenges. To reduce the shortcomings in conventional vaccinology, recombinant DNA technologies have been employed for second-generation vaccine design. Highly pure antigenic components or strains that have been rationally attenuated, for example, pertussis toxins, have been developed; however, vaccines produced using this method require several years of development [
8,
9]. In other instances, such as the development of a universal meningococcal B vaccine, the conventional empirical approach was insufficient owing to the large number of strains and the various side effects. This issue was solved by the pan-genomic method, which compares sequences from multiple strains. Proteome of serogroup B strain MC58 was investigated through various in silico methods to find possible target vaccination antigens. The genomic method also speeds up the process of vaccine development as we see in the case of mRNA vaccines developed for COVID-19. This will help in future to develop same vaccine for intracellular bacterial pathogens. The emergence of the “genomics era” has prompted a paradigm shift in vaccine development. Genome mining coupled with functional and structural genomics research has facilitated rapid antigen identification leading to third-generation vaccine development. This review focuses on the more advanced genomics-based approaches (pathogen genome and host genome) for vaccine development to treat various intracellular bacterial pathogen. We also highlight recent advancements in synthetic biology that aid vaccine design and development to address the issue of emerging intracellular infections.
Table 1.
Summary of intracellular bacteria, associated illnesses, host cells, and incubation period.
Table 1.
Summary of intracellular bacteria, associated illnesses, host cells, and incubation period.
| Bacteria | Illness | Intracellular Lifestyles | Bacterial Factors | Host Cells Localization | Incubation Period | Reference |
|---|
L. pneumophila
Non-spore forming, Gram-negative | Legionnaire’s disease | Intravacuolar
pH 6 | Type IV secretion system | Macrophages | 2–10 days | [10] |
M. tuberculosis
Non-spore forming, Acid Fast | Tuberculosis | Intravacuolar
pH 6.4 | Type VII secretion system | Macrophage, Cytosol, Phagosome | 4–6 weeks | [11] |
S. typhi
Rod-shaped, Gram-negative | Typhoid fever | Intravacuolar 5 | Type III secretion system | Macrophages | 10–14 days | [12] |
Brucella spp.
Gram-negative coccobacilli | Brucellosis, High fever | Intravacuolar 4 | Type IV secretion system | Neutrophils, Vacuole | 2–4 weeks | [13] |
Listeria monocytogenes (L. monocytogenes)
Gram-positive Bacillus | Listeriosis | Intracytosolic | In1A, In1B | Epithelial cells, Cytosol | 1–2 weeks | [14] |
Rickettsia rickettsii (R. rickettsii)
Gram-indeterminant coccobacilli | Rocky Mountain spotted fever | Intravacuolar | Type IV secretion system | Monocytes/Macrophages | 2–5 days | [15] |
S. flexneri
Non-spore forming, Gram-negative | Enteric disease | Intracytosolic | Type III secretion system | M Cells and Macrophages | 12–96 h | [16] |
M. leprae
Acid Fast
Bacillus | Leprosy | Intravacuolar | Type VII secretion system | Schwann cells (SCs) | 4–5 years | [17] |
S. typhimurium
Non-spore forming, Gram-negative | Indigestion, food poisoning | Intravacuolar
pH 5 | Type III secretion system | Macrophage, Vacuole | 12–72 h | [18] |
Chlamydia spp.
Gram-indeterminant | Genital and ocular infections | Intravacuolar
pH ¼ 7.25 | Type III secretion system | Genital epithelium and conjunctiva, Vacuole, | 1–2 weeks | [19] |
S. aureus
Gram-positive | Dermal infection, osteomyelitis, mastitis | Intravascular | Type IV secretion system | Epithelial Cells, Osteoblast, Endosome, Cytosol | 16–18 h | [20] |
S. enterica
Gram-negative | Paratyphoid and typhoid | Intravacuolar | Type III secretion system | Macrophage, Modified phagosome, Vacuole | 12–72 h | [21] |
Table 2.
Vaccine types for immunization against different bacterial infections (reprinted from Osterloh, 2022) [
22].
Table 2.
Vaccine types for immunization against different bacterial infections (reprinted from Osterloh, 2022) [
22].
| Vaccine Type | Vaccine (Target Bacteria) | Vaccine Status | Reference |
|---|
| WCA | Q-Vax (C. burnetiid)
(Dukoral, Shanchol (V. cholerae)
R. rickettsia
R. prowazekii
O. tsutsugamushi | Licensed
Licensed
Experimental
Experimental
Experimental | [23]
[24]
[25]
[26]
[25] |
| LAVs | BCG (M. tuberculosis)
S. enterica spp.
BioThrax (B. anthracis)
LVS (F. tularensis)
Vaxchora (V. cholerae)
O. tsutsugamushi
R. prowazekii | Licensed
Licensed
Licensed
Licensed
Licensed
Experimental
Experimental | [27]
[28]
[29]
[7]
[30]
[31]
[32] |
| Live recombinant bacteria | M. tuberculosis
R. rickettsii
C. burnetii
S. aureus | Experimental
Experimental
Experimental
Experimental | [33]
[34]
[35]
[36] |
| subunit vaccines | dTAP combined vaccine (C. tetani)
dTAP combined vaccine (B. pertussis)
Trumenba (N. meningitidis)
rPA102 (B. anthracis)
S. aureus
Trumenba (N. meningitidis) | Licensed
Licensed
Licensed
clinical trial
Experimental
Licensed | [37]
[38]
[39]
[40]
[41]
[42] |
| Polysaccharide conjugates | PedvaxHIB, ActHIB, HibTITER (H. influenzae)
Prevnar 13, Pneumovax 23 (S. pneumoniae)
Menactra, Menveo,
Menomune (N. meningitidis) | Licensed
Licensed
Licensed | [43]
[44,45]
[46]
|
| Viral vectors | 85A antigen (M. tuberculosis) | Licensed | [47] |
| Bacterial vectors | H. pylori (S. typhimurium) encoding urease A and B subunits
Y. enterolica encoding bacterioferritin (B. abortus)
L. monocytogenes encoding antigen 85 complex and MPB7MpT51 antigen (M. tuberculosis) | Experimental
Experimental
Experimental | [48]
[49]
[50] |
| Plasmid DNA | M. tuberculosis (hsp65 from M. leprae)
B. anthracis (PA antigen) | Experimental
Experimental | [51]
[52] |
| BGs | Y. pestis
S. typhimurium enteritides (BGs expressing flagellin)
BGs carrying DNA for N. ghonorhea antigens | Experimental
Experimental
Experimental | [22]
[53]
[54] |
| OMVs | Bexsero/4CMenB, VA-MENGOC-BC, MeNZB, MenBVac (N. meningitidis serogroup B)
B. pertussis
BCG
C. trachomatis
V. cholerae
M. smegmatis
T. pallidum | Licensed
Experimental
Experimental
Experimental
Experimental
Experimental
Experimental | [55,56,57]
[22]
[22]
[58]
[58]
[58]
[58] |
3. Genomics Revolution
The beginning of genomics can be traced to the 1970s, when DNA sequencing technology was first developed. However, the “genomics era” truly began in the late 1990s with the sequencing of the
H. influenzae genome [
59]. Thereafter, the emergence of advanced technologies made it possible to swiftly sequence a genome, thus leading to the conventionalization of whole-genome sequencing [
60]. For all major human pathogens, at least one genome sequence is available. As of July 2022, 190,825 (88.2%) bacterial genomes were assigned a “completed” status, inclusive of closed genomes and whole-genome shotgun sequences, while the sequencing of 19,217 (8.9%) genomes is ongoing (
https://gold.jgi.doe.gov/index (accessed on 12 August 2022). The sequencing status of the remaining genomes include 4907 (2.3%) in draft, 966 (0.4%) awaiting samples, 389 (0.2%) undergoing targeted gene sequencing, and 12,895 completed viral genomes (
https://gold.jgi.doe.gov/index (accessed on 12 August 2022). The sequencing of bacterial pathogens (~4000 genes) has helped identify all antigens that can be potential drug targets. Viral pathogens have less than 10 genes; however, their genomics can be used to identify the variability between different isolates. Furthermore, the role of host genetic factors in infectious diseases is also very important. Therefore, the availability of whole human genome sequences and various ongoing human genome projects (
http://www.1000genomes.org/ (accessed on 14 August 2022) are valuable assets that facilitate the identification of numerous possible vaccine and drug targets [
61,
62]. Genome-based approaches may potentially help identify 10–100 times more candidates in 1 to 2 years compared to conventional methods over the same period [
63]. Advancements in long reading sequencing technologies will facilitate the systematic assembly of diploid genomes, revolutionizing genomics by presenting the entire range of human genetic variation, covering some unaccounted heritability, and uncovering novel disease processes. Aside from this, genome-based vaccine projects enhance our understanding of pathogenesis, epidemiology, physiology, and the function of microbial proteins [
64]. Causative agents responsible for disease outbreaks can be identified by metagenomics (the evaluation of all genetic information extracted directly from a sample) [
65,
66]. The complete genome sequence identification of organisms serves as a starting point for the screening process of target molecules using high-throughput sequencing approaches, as shown in
Table 3 [
60,
67]. The type of pathogen being screened determines the screening methods used for vaccine design; however, vaccine design is also based on several widely accepted principles and essential criteria for therapeutics. These criteria include the need for targets that (a) are expressed and accessible to the host immune system, or a therapeutic agent during disease; (b) are conserved genetically; (c) exhibit survival and pathogenesis significance; and (d) demonstrate no similarity to various other attributes of the host. Previous decades have seen significant advances in the application of genomic technology that has led to new vaccine development strategies against major human pathogenic bacteria such as intracellular bacterial pathogens (
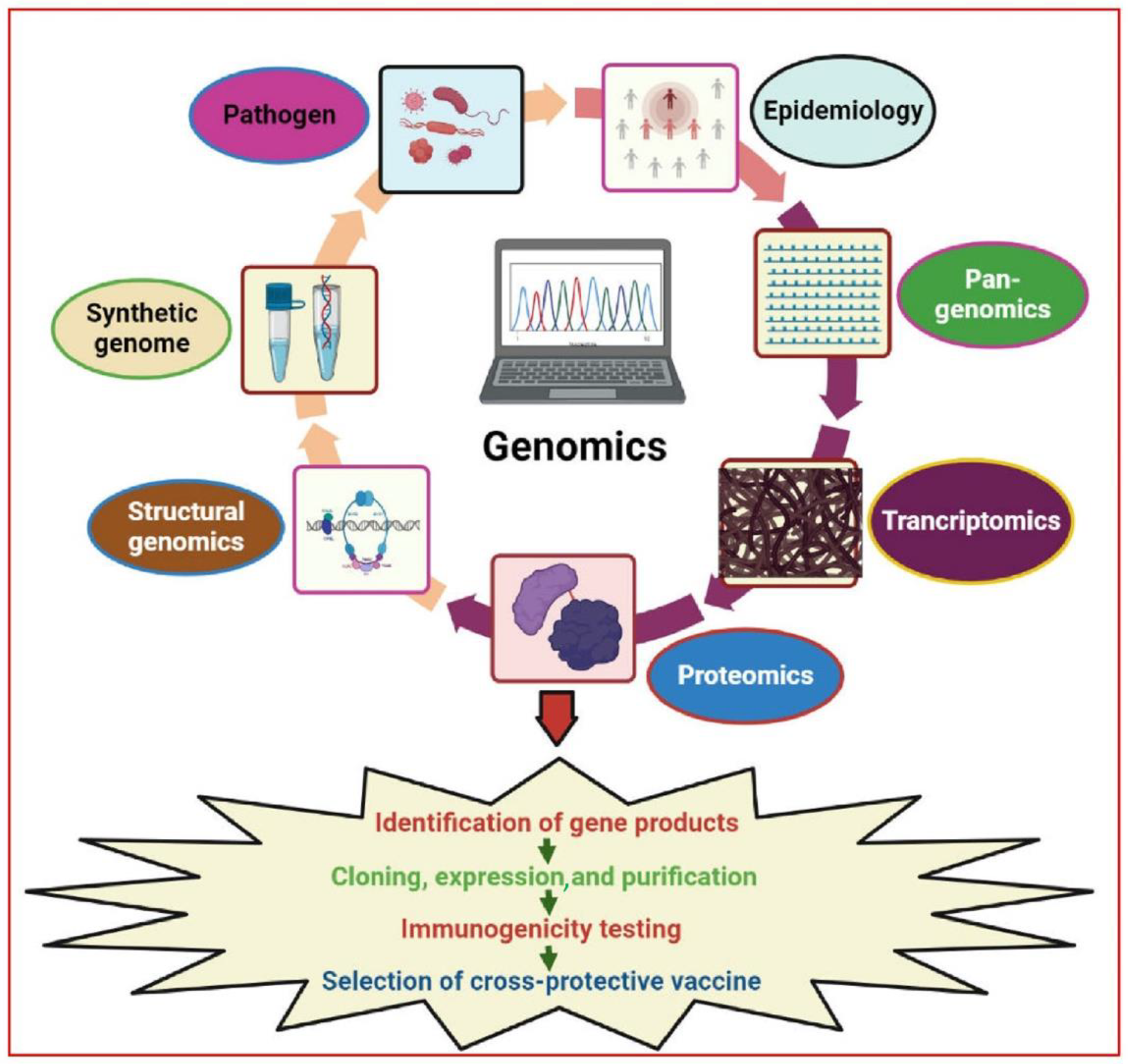
Figure 1) [
22,
68,
69,
70,
71,
72].
5. An In-Silico Approach: Reverse Vaccinology (RV)
Genome mining is a major approach used in reverse vaccinology (RV). It uses the sequences of various organisms of interest, such as viruses, bacteria, or parasitic pathogens, instead of cells as starting material for the identification of new antigens that can subsequently be verified by experiments (
Table 3) [
80]. In this method, the pathogen is sequenced and analyzed in silico to identify genes that are most likely to encode surface-localized proteins or those that have homologies with known bacterial components involved in bacterial pathogenesis and virulence. The gene of interest (GOI) is then isolated, cloned, expressed as a recombinant protein, purified, and used in animal model testing to elucidate the capacity of the gene to confer defense against the tested pathogen (
Figure 2). Alternately, in the absence of an in vivo model, in vitro approaches that elucidate vaccine efficacy in humans, such as opsonophagocytosis and serum bactericidal activity assays, may also be carried out [
81,
82]. Pizza and colleagues in collaboration with The Institute for Genomic Research (TIGR) first applied the RV approach to examine the intracellular bacteria belonging to
N. meningitidis serogroup B (MenB), which is responsible for 50% of meningococcal meningitis cases worldwide [
83,
84]. Based on the chemical composition of polysaccharide capsules,
N. meningitidis was identified as having 13 serogroups. Only five serogroups (A, B, C, Y, and W135) have been linked to meningococcal meningitis and sepsis. Capsular polysaccharides have been used in vaccines against serogroups A, C, Y, and W135 in both adults and infants. Interestingly, it was found that when the bacterial polysaccharide was conjugated with a carrier protein, a strong T-cell-dependent immune response was evoked following vaccination that provided long-term protection. However, such an immune response was not observed for serogroup B bacteria. Initial attempts to develop a meningococcal B vaccine by conventional methods were unsuccessful. Although there were several reasons for this failure, two primary reasons are apparent. First, meningococcal B capsular polysaccharides (CPS) (a polymer of α (2-8)-linked N-acetylneuraminic acid) had a high similarity to components of human tissues, which resulted in poor immunogenicity in humans, often stimulating autoimmune responses. Secondly, as protein-based vaccines are highly antigen-specific, they only provide defense against a very small number of strains [
85]. Briefly, in RV, the virulent strain of
N. meningitides MC58 was completely sequenced, and an in-silico method was used to identify 600 genes that encoded surface-exposed proteins [
83]. Out of these 600 genes, 350 were expressed successfully in
E. coli, and were subsequently used to examine immunogenicity in mice [
83]. Enzyme-linked immune sorbent assay (ELISA), fluorescence-activated cell sorting (FACS), and immunoblot analysis were performed using the immune serum of the mice to examine surface-exposed localization [
83]. These experiments identified 90 previously unknown surface-localized proteins. Bactericidal assays and/or passive protection in infant rat assays were subsequently carried out to identify 30 out of the 90 novel proteins that triggered the production of antibodies, which could eradicate the bacteria in vitro. Clinical trials were subsequently carried out using five antigens that had a high degree of conservation in multiple
N. meningitides strains. Additionally, the five antigens could also be purified with relative ease. The findings of this study led to the development of 5CVMB, a recently released universal vaccine against
N. meningitidis, a “cocktail” of five antigens discovered by RV (fHBP, NadA, GNA2132, GNA1030, and GNA2091) [
86].
The RV procedure has also been used to examine many other human bacterial pathogens [
83]. The antigens that were identified have been successfully advanced to the clinical and development phases [
87,
88,
89,
90,
91,
92]. The list of bacteria includes
Streptococcus species (
S. agalactiae, S. pyogenes, S. pneumoniae),
B. anthracis,
Porphyromonas gingivalis (
P. gingivalis),
M. tuberculosis,
H. pylori, and
Chlamydia pneumoniae (
C. pneumoniae) (
Table 4) [
87,
88,
89,
90,
91,
93,
94]. Interestingly, the investigation of 8 group B
streptococcus genomes led to the discovery of 312 surface proteins, of which 4 proteins progressed to vaccine development and were later found to be effective against all subtypes [
95]. For group A
Streptococcus (GAS) bacteria, a vaccine containing a small number of bacterial proteins has also been produced [
96]. In both instances, the RV approach was crucial for ensuring that the chosen antigens had no similarity to proteins encoded by the human genome. The sharing of epitopes between host antigens and streptococcal bacteria is referred to as molecular mimicry [
97]. Damian was the first to propose the word “molecular mimicry’ from an evolutionary perspective [
98]. He postulated that antigenic sharing between microbes and host tissue could be seen as a way for pathogens to evade the host’s immune response [
98]. In this context, the very first example was provided by Zabriskie in GAS [
99]. The hallmark of rheumatic fever pathogenesis is molecular mimicry, where the GAS carbohydrate epitope, N-acetylglucosamine, and
streptococcal M protein structurally mimic cardiac myosin in human disease [
100]. In animal models immunized with the M protein of streptococcus and cardiac myosin, pancarditis was observed [
100]. GAS vaccine candidate screening is based on the conserved M epitope, N terminal M peptides, the cell surface, and secreted proteins [
101]. Genetics and proteomics can be used to precisely formulate the antigen/epitope composition of novel vaccines. However, a vaccine is still not available, and various options are being examined for vaccine development. Vaccines based on the M protein (purified M protein, StreptInCor, 30 valent HVR StreptAnova, J8/J14/p145) are in phase I clinical trials, while the 23 valent HVR vaccine candidate has moved on to clinical trial phase II [
101]. Comparative genomics is another approach that can be used to identify possible candidate GAS vaccines.
Protein-based vaccines are also being developed using a genome-based strategy to protect against emerging infectious diseases caused by
Streptococcus pneumoniae (
S.
pneumoniae) and antibiotic-resistant
S. aureus, C. pneumoniae, etc. Additionally, a vaccine against
Chlamydia has been developed using this strategy [
102].
Chlamydia pneumoniae is a pneumonia-causing species of intracellular bacteria. The absence of an effective pneumonia vaccine has allowed the bacteria to develop substantial antibiotic resistance. Researchers have attempted to utilize proteomics and RV techniques with the aim of develo** a multi-epitope vaccine [
103]. The efficacy of the vaccine was validated using immunology and bioinformatics-based integrative pipeline (a series of software algorithms to generate processes and data) approaches. The vaccine developed by this method exhibited sustained binding to TLR4, MHCI, and MHCII receptors, as well as the capacity to trigger the host immune response. Further scientific evaluation of vaccine candidates will enable us to efficiently combat endemic diseases caused by intracellular bacterial pathogens. Most vaccines designed using the RV approach are in the discovery phase, while some candidates have reached phase I and II clinical trials (
Table 4). The main disadvantages associated with protein-based vaccines include low protein abundance and/or solubility. Moreover, proteins that are only expressed in vivo may also be difficult to identify.
The RV method has also been utilized to examine
M. tuberculosis, a bacterium which causes tuberculosis (TB), the thirteenth leading cause of death (1.6 million) and second leading infectious killer (COVID-19 being the first) worldwide (
https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 15 November 2022). Low efficacy of the
Bacillus Calmette–Guérin (BCG) vaccine against pulmonary TB, coupled with the emergence of multidrug-resistant TB, warrants the need for novel TB vaccines. The BCG vaccine is not universally approved. It is not administered in many developed countries; however, it is still used in underdeveloped and develo** countries. The status of TB in different countries, in relation to BCG vaccination is available online (
http://www.bcgatlas.org/ (accessed on 16 November 2022). Anti-mycobacterium vaccines are urgently required. With respect to this, several web-based RV programs (MycobacRv, Violin, VaxiJen, and MtbVeb) have been developed to aid the scientific community. MycobacRv is a database of potential mycobacterial adhesin vaccine candidates identified from 22 mycobacterial strains. The database also provides detailed analysis of predicted adhesins/adhesin-like and extracellular/surface-localized proteins, which may help in the creation of epitope-based mycobacterial vaccines [
104]. Another web-based database called the Vaccine Investigation and Online Information Network (VIOLIN) combines mined vaccine literature with data curation and storage. Additionally, it offers a framework that can predict possible targets for vaccines against different pathogenic invaders [
105]. Recently a study analyzed the proteome of
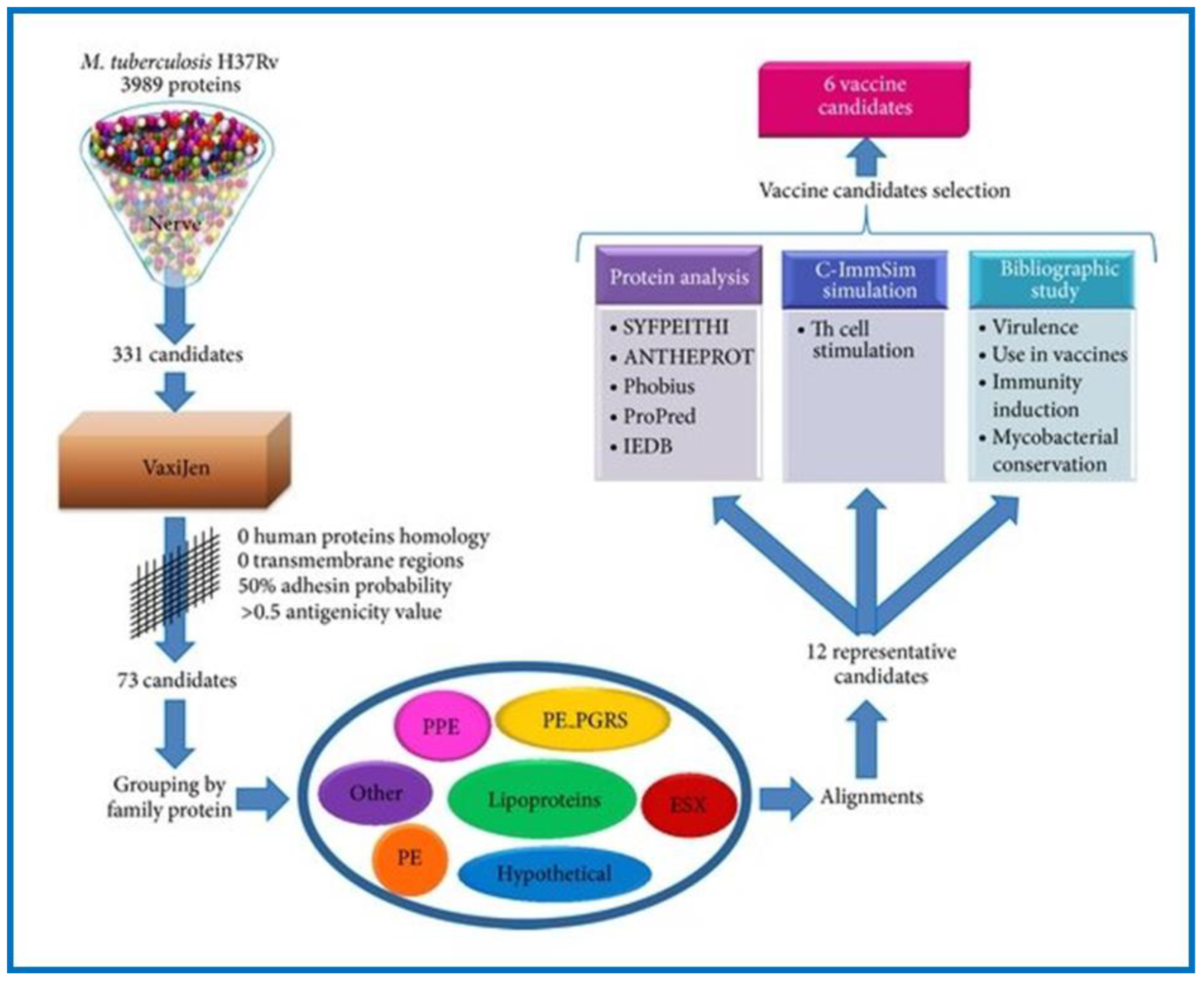
M. tuberculosis H37Rv using a prediction software called the New Enhanced Reverse Vaccinology Environment (NERVE) to identify vaccine targets. NERVE identified 331 proteins, which were then further analyzed by VaxiJen software (
Figure 3). Specific filter parameters (adhesin probability value 50%, antigenicity value ≥0.5, and no homology to human proteins or transmembrane sections) were used to identify 73 antigens. Further search refinements involving detailed literature and protein analysis revealed six novel vaccine candidates (EsxL, PE26, PPE65, PE_PGRS49, PBP1, and Erp), which were subsequently approved for TB vaccine development [
106]. Clinical trial data for PE_PGRS49 (a DNA vaccine with a protein-related member Rv1818c) have been reported [
107], while data for the remaining five have not. The inclusion of PE_PGRS49 in existing BCG vaccines may enhance their efficacy [
106]. Recently, the RV strategy was successfully applied to many other intracellular bacterial pathogens for vaccine development, including
Brucella spp.,
C. pneumoniae,
R. prowazekii,
Anaplasma spp.,
Ehrlichia spp.,
S. pyogenes, etc., [
25,
103,
108,
109,
110,
111,
112,
113,
114].
The fundamental limitation of the RV strategy is that it only examines one single strain within a species, and does not address issues that may arise owing to genetic diversity. This risk became apparent when a comparison of genome sequences from several strains of
S. agalactiae were carried out. Eighty percent of the
S. agalactiae core genome is composed of genes, with each new genome displaying ~18% variation. This finding served as inspiration for the development of a “universal anti-
S. agalactiae” vaccine. This vaccine was composed of four antigens, with one antigen being present in all strains, and this combination provided protection against all strains. The evolution of sequencing approaches provided researchers with an enriched bacterial genome sequence. This opened a new area of vaccine development known as pan-genomics. The comparative or pan-genomic method was viewed as the development of classical RV. This method facilitated the development of universal vaccines based on a core genome shared by all strains, which would be effective against various strains of the same species [
115,
116,
117,
118].
6. Pan-Genomics Analysis or Comparative Genomics
Following publication of the first bacterial genome sequence, technological advancements have led to the elucidation of numerous complete genome sequences. The genomic databases now have more than 250,000 bacterial genomes, a result of intensive work and more efficient sequencing methods [
119]. The immense genomic variation among individuals of the same species is one of the most profound discoveries that have been made. Many mechanisms govern genetic diversity, such as genetic drift, natural selection, and gene flow due to mobile elements, transposons, and non-random mating [
120]. Subtractive hybridization and comparative genome hybridization (CGH) techniques have shown that intraspecies genetic diversity is as significant as interspecies diversity [
121] Genomic comparisons of multiple isolates of bacterial pathogens and closely linked pathogens are important for the scientific community, as knowledge regarding genome size, gene content, conservation, and gene diversity in various strains and disease conditions has profound implications on vaccine development. Further improvements in sequencing technology facilitate comparisons among multiple complete genomes. Pan-genomics (an advancement of classical reverse vaccinology) is utilized to make comparisons amongst multiple strains of bacteria.
The term “pan-genome” was first used by Tettelin et al. in 2005 (
Figure 1) [
122]. It was explained as the entire gene repertoire of a particular species. By comparing the complete genomes of eight different strains of
S. agalactiae (group B
streptococcus; GBS), representing the genetic variety of the species, intraspecies heterogeneity was demonstrated. The first application of pan-genomics was carried out to design a universal vaccine against GBS [
114]. Computational techniques were used to predict 589 genes encoding surface-associated proteins, 396 of which were core genes, while the remaining 193 genes were lacking in at least one strain. Potential antigens were subsequently selected and expressed as recombinant proteins. These recombinant proteins were purified and their anti-GBS potential was evaluated. Only four of the candidate antigens elicited immunity in animal models. Out of these four, one was a component of the core genome; however, the antigen was unable to provide universal protection against GBS. Therefore, a combination of the four antigens should be included in the final vaccine formulation. This example clearly illustrates the need to understand pan-genomics. Phase I testing for a GBS vaccination is currently underway (
http://clinicaltrials.gov/ct2/show/NCT01193920 (accessed on 17 November 2022). For GAS, comparative genomics was employed to search for a global vaccine candidate using 2083 globally sampled GAS genomes [
123]. The study reported 290 related genomic phylogroups from 22 countries, emphasizing the difficulties of develo** vaccines that have universal applicability.
Machine learning has facilitated the pan-genomic sequencing of 1595 strains of intracellular
M. tuberculosis (Mtb) bacteria [
124]. Amongst these strains, 946 bacteria were resistant to anti-tuberculosis medicine (isoniazid and rifampicin) [
124]. A pan-genomic analysis of 36 intracellular Mtb isolates identified 67 super core genes (SCGs), of which 28 were very important and reflected phenotypic generality [
105]. Most SCGs have been shown to play significant roles in Mtb pathogenicity and code proline-glutamate/proline-proline-glutamate (PE/PPE), virulence factors (VFs), antigens, and transposases [
125,
126,
127]. The proteins are the multifunctional immune modulators. PPE19 is highly expressed in macrophages (two copies/Mtb strain), which promotes Mtb intracellular survival [
127]. Another SCG, phospholipase C (plcC; three copies per Mtb strain) aids Mtb escape from phagosomal vacuoles and hence degradation [
126]. The study carried out a pan-genome investigation of Mtb to determine which genome was primary, secondary, phenotype-general, phenotype-specific, and interconvert during evolution [
126]. This study provides a novel research paradigm for investigating organisms using a pan-genomic approach as well as laying an important theoretical foundation for TB research.
In a recent study, a silicon-based multi-epitope vaccine was designed based on 200 Mtb genomes [
128]. Two vaccine candidates, ESA-6-like proteins and diacylglycerol acyltransferase were generated from multi-epitope map**. These two vaccines exhibited rigorous van der Waals and electrostatic binding forces, stable dynamics, and high binding affinities for several immunological receptors. The researchers noticed remarkable primary, secondary, and tertiary immune responses to the antigens in addition to a raised interleukin and interferon count [
129]. In conclusion, the proposed vaccines appear to be viable candidates for further evaluation. Their true biological value as agents that prevent drug-resistant
M. tuberculosis infections must be determined, along with their respective modes of action.
Salmonella typhi,
S. typhimurium, and
S. enterica cause typhoid, paratyphoid fever, and gastroenteritis. Novel antimicrobial agents are urgently needed to treat these conditions. Pan-genome analysis estimates that
Salmonella has an “open” pan-genome that has 10,775 gene families. There are 2847 core gene families (CGFs), 4657 dispensable gene families (DGFs), and 3271 strain-specific gene families (SSGFs). The study concluded that essential core gene families (E-CGFs) may serve as important targets for the development of novel antimicrobial drugs [
130]. Another pan-genome study, conducted in Brazil, revealed that while the pan-genomes of
S. typhimurium under study were open, they especially tended to close for ST313 strains [
131].
Legionella pneumophila, an intracellular bacterium linked to Legionnaire’s disease, was found to infect ~90% of patients [
132,
133,
134].
Legionella pneumophila is resistant to many drugs. Genomic approaches have been considered for the development of new vaccines. In a recent study, subtractive proteomics (pathogen genes necessary for its survival, yet absent in the host, are “subtracted”) and immunoinformatic tools were used to design a highly immunogenic vaccine containing five proteins (Q5ZVG4, Q5ZRZ1, Q5ZWE6, Q5ZT09, and Q5ZUZ8) against
L. pneumophila. The proposed vaccine elicited a host immune response against
L. pneumophila [
134]. However, to demonstrate the protective immunological efficacy of the vaccine, experiment-based studies are strongly advised. The vaccine should be applied to animal models followed by human clinical trials.
Rickettsia rickettsii (an intracellular coccobacillus bacterium) causes Rocky Mountain spotted fever (RMSF). Docking analysis, reverse vaccinology for protein identification, and pan-genomics were used to analyze 47
Rickettsia genomes. Researchers found 90–100% homology among the four
Rickettsia groups. In addition, eight protein types were also identified in this research to support polyvalent vaccine development against
Rickettsia, while nine candidates were identified as drug targets [
135]. Although the study predicted that polyvalent vaccines would interact with the majority of microorganisms in this vast group, potentially leading to a vaccine, predictive data must also be supported with in vivo and in vitro data.
Shigellosis is a disease commonly found in children from develo** countries. It is caused by the
Shigella flexneri (
S. flexneri) bacterium. Researchers have compared the virulence signatures and genomic characteristics of the of
S. flexneri stereotype 3b genome, SFL 1520, with commonly found genomes of
S. flexneri to identify a link for vaccine development [
136]. The study reported that SFL1520 shared significant similarities with other
S. flexneri serotypes in the phylogenetic analysis based on core genes, but there were also notable differences in SFL1520’s accessory genes. The discovery of a substantial number of distinct genes in SFL1520 suggests extensive horizontal gene acquisition in a relatively short amount of time. Major virulence characteristics of SFL1520, such as serotype conversion and multidrug resistance, were created by these acquired genes, which will further help in vaccine design. A deeper understanding of strain variability could serve as a foundation for learning more about variations in pathogenesis and pathogen–host interactions that can translate to healthcare benefits. The
Chlamydia group consists of a single genus, containing the species
Chlamidia trachomatis (
C. trachomatis)
, a pathogenic intracellular microorganism involved in sexually transmitted diseases causing “genetic infection” [
137]. In a pan-genomic analysis of 16 genomes examining a combination of core genomes (small and large), the stability of
Chlamydia genomes was verified, and various distinct genome characteristics were discovered [
138]. The presence of a conserved core genome (large) and evolvable variable (small) genome balance the selective pressure towards genome reduction and the requirement for adaptation to evade host immunity.
Gastritis and stomach ulcers are caused by the Gram-negative bacterium
H. pylori. One to two percent of infected individuals may develop stomach cancer.
Helicobacter pylori can be transmitted from person to person through saliva, or through contaminated food or water. Additionally, 80% of infected people have no symptoms, and bacterial resistance to medication has increased over the last decade. To control
H. pylori infections, vaccine-based prevention has been prioritized rather than subsequent antibiotic treatment. The RV strategy in combination with pan-genomic analysis was carried out in a study to analyze 39
H. pylori isolates [
139]. A total of 28 non-host homologous proteins were identified as common therapeutic targets for future vaccine development [
139].
Table 5 shows the details of antigens or approved vaccines against bacterial pathogens that were identified by the pan-genomics RV method.
7. Antigen Prioritization
Another primary disadvantage of the RV method is that it is often expensive and labor-intensive. High-throughput screening of countless antigens also makes this approach time-consuming. RV findings have demonstrated that when seeking new vaccine candidates, surface-exposed bacterial proteins that may trigger a strong immune response should be taken into special consideration. More stringent in silico screening measures and the application of new experiment methodologies should be prioritized to meet these requirements. A study examining the relationship between Tourette syndrome (TS) (a neurodevelopmental disorder beginning in childhood or adolescence) and
S. pyogenes (or Group A streptococcus (GAS) antigen, including subsets of M proteins, streptolysin O (SLO), streptokinase A, and C5a peptidase precursors)-induced immune response implemented this strategy [
118]. Many studies have reported a highly evolved relationship between TS and streptococcal infection [
147,
148,
149]. Stringent in silico analysis was used to filter gene sequences to predict surface-associated proteins that had canonical motif-associated surface-exposed proteins. These surface-exposed proteins contained a lipoprotein signature, host cell binding domains (RGD), leader peptides, and outer membrane anchoring motifs. The screened protein coding genes were cloned, purified, and used for GAS-specific protein microarray analysis. The detailed protocol has also been reported [
118]. The array proteins were then further examined using human sera from asymptomatic isolates, symptomatic isolates of pharyngitis, and TS isolates. This provided the first proof that the sera from TS patients had immunological profiles similar to patients who exhibit a strong, specific, and broad immune response. The same strategy may be applied to identify novel vaccine candidates. This in silico method of canonical surface motif screening minimizes the number of genes that have to be cloned and the number of proteins that have to be purified, ultimately reducing the quantity of pathogenic antigens that need to be examined to identify high antigenicity.
Another approach prioritizes candidate antigens for vaccine development based on immunogenicity. This approach has been used with respect to several intracellular bacterial pathogens [
146,
150,
151]. In this method, extensive libraries of peptides expressed on the surface of
E. coli are created and analyzed using human serum from infected or recuperating patients. The main advantage of this approach is that it is a simple serum analysis method; however, a disadvantage is that the proteins are not expressed because they do not fold properly. Although in this process, some antigens might be overlooked when comprehensive cloning, expression, and purification are not possible, it is both cost- and time-efficient. The proteomics-based strategy, which entails the proteolytic “shaving” of live bacterial cells (to identify proteins that are easily accessible to host immune functions), is another experimental means to prioritize specific antigens from a list of in silico selected surface proteins. Briefly, proteases are used to selectively digest bulging proteins from whole cells that have been identified by the mass spectrometry analysis of released peptides and genome-based antigen analysis. Rodriguez-Ortega et al. also used the same method for GAS surface proteome analysis and identified 70 proteins [
146]. Known protective antigens were excluded from the selection, while new antigens were expressed, purified, and later used in mice immunization experiments. This technique has also been carried out to examine a wide range of bacterial pathogens, including M.
tuberculosis, S.
dysenteriae,
F. tularens, and
B. anthracis [
152,
153,
154,
155,
156,
157].
9. Transcriptomics: Expression Profile Identifies Potential Vaccines
Transcriptomics offers a comprehensive perspective of a pathogen’s full transcriptional activity and allows the comparison of gene (RNA transcript) expression under various growth and environmental settings. Knowing which genes are overexpressed in vivo during infection is crucial for the discovery of vaccine antigens, potentially serving as candidates for prospective vaccines. Owing to whole-genome sequencing, complete microbial genome sequences are now more easily accessible, which has facilitated their usage in various vaccine development strategies.
To identify promising MenB vaccine candidates, Grifantini and colleagues conducted microarray-based transcriptional profiling of the intracellular bacterium,
N. meningitidis [
160]. In this investigation, bacteria were cultured with human epithelial cells. Adhered bacteria were retrieved, and total RNA was extracted over a period. RNA was also extracted from non-adherent bacteria that were cultured in the absence of epithelial cells. DNA microarrays that contained the complete repertoire of the MenB gene amplified by PCR were compared. Twelve proteins that were found to be highly stimulated during adhesion were purified. The purified proteins were subsequently utilized to generate antisera in mice. A total of five sera demonstrated antibactericidal properties. Furthermore, subsequent transcriptomic investigations revealed that 48 differentially expressed genes (DEGs) were also present during the interaction of
N. meningitidis with blood–brain barrier endothelial cells [
161]. Of the 48 genes, 41 were overexpressed, while 7 were downregulated. The majority of DEGs were involved in metabolism, transport, protein production, and pathogenicity. Transcriptional profiling of MenB has identified genes that were predicted to encode for pathogenesis-related proteins [
160]. In these studies, various experiment conditions, such as human serum and endothelial cell exposure, iron restriction, oxygen starvation, etc., were applied [
160,
162]. Several new genes that are likely to encode proteins implicated in the disease process were identified, along with their potential function. For example, the ferric uptake regulator (Fur) and iron-regulated operon of
N. meningitidis are necessary for oxidative stress defense. Furthermore, fumarate and nitrate reductase regulator (FNR) proteins and FNR-associated proteins carry out carbohydrate fermentation, which is necessary for MenB survival in regions of the host where oxygen is limited.
The transcriptomic comparison of a clinical isolate of
S. aureus, UAMS-1, with a laboratory strain, RN6390, revealed significant differences in the expression of genes encoding surface proteins (elevated in UAMS-1) and genes encoding proteins involved in exotoxin production (low in UAMS-1, having exotoxin 2 or 3), shedding light on pathogenesis, and emphasizing the value of investigating clinically appropriate strains for vaccine development [
163,
164]. With the discovery of many advanced sequencing technologies (RNA sequencing), the complete transcriptome of pathogenic bacteria, containing complete gene expression data, can be harnessed by researchers. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis tool enables systematic analysis of gene function, which may shed light on bacterial pathogenesis. Transcriptomic profiling of
M. tuberculosis in an early tuberculosis BALB/c and SCIF immunocompetent mouse model showed that the activation of 67 genes in mice lungs correlated with the activation of the host immune response [
165,
166]. The benefits of transcriptome analysis for vaccine development greatly increased following technological advancements in the in vivo isolation of microbial RNA from tissues [
167,
168].
The rapid sequencing of cDNA and quantification of sequence reads, made possible by the increasing availability of microarrays (for example, for 39 different pathogens arrays are available freely from J. Craig Venter Institute: Pathogen Functional Genomics Resource Center;
http://pfgrc.jcvi.org/index.php/microarray/available microarrays.html (accessed on 25 September 2022), should allow for further transcriptome-based vaccine development [
169,
170].
The use of next-generation sequencing (NGS) to investigate complex processes, such as bacterial infections and diseases, has greatly advanced our understanding of crucial components at both the genomic and transcriptome level, giving light to the changes within pathogens in response to therapeutic interventions [
171,
172]. For example, NGS has been used to identify the mutation and recombination events that enabled 240 multidrug-resistant strains of
S. pneumoniae to evolve [
171]. A phylogenetic reconstruction of the genesis and global distribution of these strains was made possible by the identification of more than 700 recombination events and 57,736 single-nucleotide polymorphisms (SNPs) in total. Similarly, use of RNA-Seq (to measure the complete transcriptome quantitively) to analyze the transcriptome of
H. pylori RNA, revealed a multitude of complexities, and the detection of hundreds of transcriptional start sites [
173]. These state-of-the-art technologies have helped scientists obtain sequencing information of bacteria in a short period of time, facilitating rapid vaccine design.
10. Proteomics: A Genomic Complement for Vaccine Development
Another functional genomics tool is proteomics, the examination of proteins expressed in a cell. It enables the varied expression of proteins to be characterized as well as their location; for example, proteins that are found outside the cell, that is, on the cell surface proteome (surfaceome), are essential for triggering immune responses [
174,
175]. Information processing (mediated by proteins on the cell surface) is necessary for communication between immune system cells and the organism. The cell surface proteome is composed of a collection of functionally diverse proteins that both promote and inhibit the ability of cells to interact with one another in the environment. Proteomics-based methods for identifying surface-associated immunogenic proteins, which can serve as vaccine candidates, have rapidly advanced in the genomic era, and are now commonly regarded as efficient technologies that complement traditional genomic methods. The availability of an ever-increasing number of whole-genome sequences has enabled the proteomic community to identify proteins of interest swiftly and accurately from various cell compartments. In order to overcome the limitations of genomics methodologies, such as the fact that (1) expression levels of mRNA do not accurately reflect the quantity of active protein in a cell, (2) gene sequences provide insufficient data on post-translational alterations, and (3) dynamic cellular processes are not defined by genomic information, proteomics has been employed in a variety of ways to uncover new vaccination candidates against a variety of human illnesses [
67,
176,
177,
178,
179,
180,
181]. For the proteomics approach, two-dimensional gel electrophoresis or liquid chromatography (LC) are used to reduce the complexity of samples by separating proteins or peptides for analysis by mass spectrometry (MS). The primary tool used for protein identification and characterization is MS, which has improved significantly in the past decade [
182]. There are three components to MS: ion sources, mass analyzers, and ion detection systems. For the analysis of proteins through MS, three parameters are considered: (1) a protein’s ability to ionize and produce gas-phase ions, (b) the separation of ions based on their mass to charge ratio, and (c) ion detection [
183]. Tandem mass spectrometry (MS/MS) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) were used to determine each peptide’s molecular mass [
67]. Peptide mass fingerprinting is a high-throughput protein identification method. The unknown protein is digested (with enzymes into smaller peptides), analyzed, and then the peak list of peptides in the unknown proteins is compared to a theoretical list of the protein mass database for identification [
184,
185]. Detailed MS procedures have previously been reported [
186]. However, this approach does not effectively identify hydrophobic proteins. Tandem mass spectrometry and two-dimensional liquid chromatography (2D-LC-MS/MS) were subsequently developed and demonstrated to be very helpful in the identification of extremely hydrophobic or basic proteins that were insufficiently expressed and had large molecular weights and excessive isoelectric points [
187,
188].
Many important methods have been developed to identify surfaceomes that have minimal cytoplasmic protein contamination. The surfaces of live bacteria are carefully digested with proteases using mass spectrometry analysis as a guiding tool. This method was also applied to bacterial culture supernatants to distinguish and analyze the secretome of bacteria [
189,
190]. Proteomic studies have been utilized to understand how the environment influences the pathophysiology of various microbes in addition to examining microbe–host interactions [
91]. With the passage of time, there has been significant advances in proteomics approaches for the screening of vaccines. A comparative approach examining differential expression parameters, such as non-virulent strains versus virulent strains, less invasive strains versus more invasive strains, or colonizing strains versus non- colonizing strains.
The most promising vaccine options for most bacterial infections are proteins that are either secreted or surface-exposed and can induce a protective immune response. In silico analyses of surface-associated proteins predict that they account for 30–40% of bacterial proteins. Rodriguez-Ortega et al. developed a novel proteomics-based method involving the precise isolation of bacterial surface proteins [
146,
152]. The proteomics approach was applied to
C. pneumoniae, an obligate intracellular bacterium causing respiratory infections.
Chlamydia pneumoniae also contributes to atherosclerosis and heart disease. Molecular characterization of the chlamydial cell surface is still limited due to the inherent difficulties of working with
C. pneumoniae and the lack of reliable methods for its genetic manipulation, leaving the processes of entry largely unexplained. Therefore, a genomic and proteomic approach was used to elucidate the organization of surface proteins. This was based on various factors, including predicative data from the reported genome, peripherally located proteins, heterologous protein expression, selected protein purification, mouse immune sera production, immunoblotting, FACS analysis (for surface antigen identification), and two-dimensional electrophoresis (2DE) [
91]. FACS-positive antigens were identified in the
chlamydial cell by mass spectrometry analysis of 2DE maps of protein extracts. The results showed that 53 positive FACS sera were identified. Out of these, immunoblotting detected 41 proteins of the correct size. Furthermore, 28 surface-exposed proteins that have potential as vaccine candidates were also identified by 2DE maps of protein extracts [
91]. The study was the first systematic attempt to explain the organization of surface proteins in
C. pneumoniae.
Several other bacteria have also been examined using the proteomics approach. For example,
Salmonella typhimurium (
Salmonella serotype (ser.)
typhimurium), a Gram-negative bacterium, has been well characterized by proteomic analysis [
191]. In 2006, analysis of the
Salmonella proteome (∼300 proteins) was performed using infected RAW 264.7 macrophage cells [
192]. Cell lysis and differential centrifugation techniques identified an excess of 2000 bacterial proteins [
192,
193]. The generated extensive dataset of the
Salmonella proteome showed substantial adaptations of the intracellular pathogen to host epithelial cells, including metabolic remodeling and differential control of virulence factors. Using the proteomics data of intracellular bacteria, the characterization of mutants can also be performed. For instance, to examine the functional roles of
Salmonella-induced filaments, the Hensel group examined the intracellular proteome of two
Salmonella mutant strains,
ΔssaV (secretion system apparatus protein, ssaV) and
ΔsseF. Furthermore, the proteome of
Salmonella mutants deficient in
ydcR (a known regulatory gene that controls the expression of virulence factor
SrfN) was also summarized [
192]. The proteomes of many other intracellular bacteria, including
S. flexneri,
L. monocytogenes,
B. pertussis,
Brucella (
B.)
abortus,
R. prowazekii, etc., were also investigated for vaccine development by many groups [
194,
195,
196,
197,
198]. This research opens the door to the development of a potent vaccine and offers a broad strategy for the design and development of vaccines against other intracellular diseases.
11. Next-Generation Epitope Map** and Vaccine Design: Structural Genomics/Vaccinology
The increasing availability of genomic sequence data led to the development of structural genomics. Structural genomics involves the advanced application of many structural biology technologies, such as X-ray crystallography, nuclear magnetic resonance (NMR), spectroscopy, and electron microscopy (EM), which are increasingly being used for vaccine development (structural vaccinology, SV) [
5]. SV is a genome-based strategy concerning the structural elucidation of immunodominant and immunosilent antigens, and the data can be used to design and develop peptide analogs of bactericidal epitopes [
199,
200]. SV has enabled antigen optimization and the large-scale industrial production of various combinations of antigens, which enforce a higher degree of immunogenicity, improved safety profiles, and enhanced protection [
201].
The advantages of the SV approach have started to become more prominent. There are numerous examples where SV has facilitated the creation of vaccines that have previously inconceivable antigen combinations. Here, we go over a few of these instances and how structural vaccinology has helped us better understand the protective epitopes of major intracellular bacterial pathogens. Capsular polysaccharides conjugated to a carrier protein are used in approved vaccines against
N. meningitidis serogroups (A, C, Y, and W135), but cannot be used against MenB, as the capsular polysaccharide is chemically identical to a human self-antigen. Recently, antigens have been identified that could possibly be used to develop vaccines against MenB. A surface-exposed lipoprotein, factor H-binding protein (fHbp), is one such protein antigen [
202,
203,
204,
205]. Although this antigen has more than 500 documented variants in its amino acid sequence, it is rather effective at eliciting protective antibodies. A study involving in vitro bactericidal assays (in which antibodies mediated the complement-dependent killing of bacteria) suggested that such variants could be divided into two or three different variant groups (referred to as variants 1, 2, and 3, henceforth) which do not elicit cross-protective immunity to one another [
202,
204]. Therefore, antigens that have specificity for all variants should be included in the vaccine design process. Although this would lead to increased complexity and an expensive production process, it would be valuable to have a single antigen that can trigger protection against each fHbp sequence variant. This was made possible by structure-based design, where epitopes from each fHbp antigenic-variant groups were designed into a single molecule. NMR and X-ray crystallography were used to identify the three-dimensional structure of fHbp, which revealed a core structure made up of two barrels joined by a shorter linker [
206,
207,
208]. The epitopes against the three antigenic variants were recognized by protective monoclonal antibodies and later identified [
202,
209]. Through a series of verification experiments, 54 molecules were designed expressed and purified. Mice were subsequently immunized with the molecules, and their sera were examined for bactericidal antibodies against MenB strains possessing fHbp 1,2, and 3 variants. Many molecules triggered a significant immune response. One molecule with high activity was chosen for additional investigation, which later demonstrated the ability to induce bactericidal antibodies against MenB strains containing fHbp variants 1, 2, and 3. The crystal structure analysis of the molecule revealed perfect conservation of the original fold, making it an excellent contender for the upcoming meningococcal vaccinations.
The SV approach has also been used for next-generation vaccine development against
Bacillus anthracis, based on its virulent and secretory proteins [
210]. Five virulent proteins have been examined in
B. anthracis to find immunogenic epitopes and develop a multi-epitope vaccine. The adjuvant Toll-like receptor 4 (TLR4), cytotoxic T lymphocytes (CTLs), helper T lymphocytes (HTLs), B cell epitopes, and linkers were added to 24 distinct subunit vaccines in various permutations and patterns. The study screened for a vaccine candidate by employing a novel strategy of SV to combat
B. anthracis, which was previously regarded as a dangerous bioterrorism agent [
210]. For the past 20 years, Willmann’s group at EMBL Hamburg has been analyzing mycobacterial proteins, utilizing high-resolution structural biology [
211]. They elucidated the cryo-EM (cryogenic electron microscopy) structure of the mycobacterial
SX-
5 type VII secretion system ‘ESX-5′ protein complex, a key component governing the virulence of mycobacterial pathogens. The structural data will aid in future vaccine design and development, as the protein complexes are a potential therapeutic target.
Structural genomics/vaccinology provides detailed insights into the structure of proteins, allowing scientists to fully consider various functional attributes. Structural genomics also provides high-quality three-dimensional structures and allows protein structure libraries to be generated, providing valuable information about wild type and mutated proteins. Structural genomics helps scientists study the molecular interaction of proteins and their folding patterns, which may further facilitate novel therapeutics. It can advance our knowledge of immune recognition mechanisms, and aid in the rational design of target epitopes that could be exploited as vaccine candidates. However, the practical applications of SV still need to be fully realized. The method cannot be accurately applied to proteins that have a low (>30%) sequence resemblance. While determining unique folds for sequences that differ from those in the Protein Data Bank, de novo protein structure prediction must be implemented. A majority of researchers who study structural genomics examine individual protein domains or subunits rather than the complete or complex protein.
12. Synthetic Genomics: The Future of Vaccine Development
Synthetic genomics is a nascent domain of synthetic biology concerned with the creation of living organisms using genetic material. In this field, genes, chromosomes, gene networks, and whole genomes are combined with the help of computational approaches for chemical DNA synthesis. Synthetic genomics aims to create new genomes that can code for specialized cells with desired traits by making significant modifications to chromosome DNA, packaging, and then introducing the material into an organism.
The history of synthetic genomics dates to 2010, when a synthetic organism was created for the first time by researchers. The J. Craig Venter team presented a multistep procedure to assemble the entire
Mycoplasma genitalium genome. This development was further enhanced by Synthetic Genomics Inc., a company that focuses on the study and commercialization of specifically constructed genomes [
212]. Synthetic genomics, which includes novel protein designs, structural analysis, and the production of recombinant vaccines for quickly evolving microbial infections, has the potential to accelerate vaccine development. David Willetts’ well-known adage, “Engineering biology to cure us, feed us, and fuel us”, is most appropriate for synthetic biology because it can affect almost every aspect of our life. The need and demand for novel innovative drugs has increased due to drug resistance.
Tuberculosis (TB), one of the deadliest risks to human health, and has pandemic potential (
https://www.tballiance.org/why-new-tb-drugs/global-pandemic (accessed on 18 November 2022). Synthetic Genomics Inc. has developed a drug against TB. The bacterium
Actinobacterium mediterranei (which produces rifamycin B and has a gene cluster for rifamycin polyketide synthase) was modified by the genetic-synthetic method to generate mutant strains that can produce 24-desmethylrifamycin B (a rifamycin analogs). The analog addresses multidrug resistance (MDR) issues involving
M. tuberculosis. Interestingly, 24-desmethylrifamycin B derivatives, namely, 24-desmethylrifampicin and 24-desmethylrifamycin, showed 10 times greater activity than rifampicin [
213]. The synthetic genomics approach may also be used to generate drug analogs that can be used to treat other dangerous types of MDR TB, such as extensively drug-resistant TB (XDR-TB) and totally drug-resistant TB (TDR-TB) [
214].
13. Conclusions and Future Direction
Through the course of human history, there have been multiple pandemics of infectious diseases, which have caused people’s viewpoints to change. The advent of the genomic era has witnessed a change in the paradigm of vaccine design from traditional culture-based approaches to high-throughput genome-based approaches. There are demands for proactive and collaborative worldwide research and vaccine development programs. To counter some of the deadlier, more widespread infectious pathogens, such as those that cause TB, meningitis, malaria, AIDS, and dengue, vaccines need to be researched and developed on an urgent basis. Where all other conventional methods fail, genomics offers an opportunity for vaccine development against widely dispersed emerging pathogens.
The genome-based revolution in vaccine development has made information regarding a pathogen’s genome, transcriptome, proteome, and immunoproteome available to researchers so that new vaccine antigens may be discovered. Furthermore, advances in our understanding of many other characteristic features of microbes, such as epidemiology, evolution, virulence gene identification, interaction of pathogen with host, intra- and interspecies complexity, and physiology, have also helped further improve vaccine development. Epidemiological and evolutionary studies examine the microevolution of strains and the identification of newly emerging clones, using various molecular biology techniques such as multilocus sequence ty** (MLST). To design a universal vaccine capable of targeting all pathogenic strains, molecular epidemiological studies consider trend changes in the population, clustering patterns (regional and worldwide), and antigenic variation among isolates. Furthermore, molecular epidemiology research can support the evaluation of a vaccines effectiveness, the incidence of vaccine escape variations, or the emergence of novel harmful variants as a result of intense selection pressure. Virulence gene identification was also made possible by complete genome sequencing of pathogenic strains. The process helped identify genetic patterns associated with disease virulence as well as the genetic elements that support immunity or a positive vaccine response. The data will help develop vaccines that have higher efficacy and specific immune responses. Recently, vaccine research and development programs are typically driven by one of these approaches or a combination of them, with the choice of technique being greatly influenced by the traits of the target pathogen. For example, the conventional RV strategy is sufficient for low-antigenic and non-cultured species, while species that have greater antigen diversity and variability may require a refined RV approach. Other approaches involving transcriptomics (RNA sequencing) and proteomics reduce the screening time of potential candidates, enabling rapid selection and evaluation of antigens.
Major barriers associated with conventional vaccine design approaches can be overcome using genomic approaches. Understanding how immunity develops is the first barrier. A possible solution may necessitate a systems biology approach involving the identification of non-humoral protection, understanding effectors and vaccines that induce cellular immunity, and clinical assessment strategies. The second barrier is host variability (age, sex, ethnic group, genetic diversity). A genomics-based solution to this includes designing population-based vaccines and carrying out diagnostic tests to predict population wide vaccine responses. The third barrier is pathogen variability (multitude of strains, varying stages of infection, different pathogen–host interactions, strain evolution). Multivalent vaccines that elicit strong immune responses in all strains (pan-genomics) and lead to the production of several neutralizing antibodies have been designed. The fourth barrier concerns vaccine safety. Vaccines may have side effects, involving autoimmune responses. Individuals may also refuse to get vaccinated owing to such concerns. To minimize safety-related issues, novel subunit and protein adjuvants may be incorporated into the design of vaccines to enhance specific immunogenicity and grant durable protection. Lastly, non-heritable factors (eating habits, alcoholism, smoking, living environment/geography, etc.) that affect vaccine efficacy are also major barriers. Genomics approaches partially resolve these issues as vaccines can be specifically designed for naïve and exposed individuals. Vitamin supplements that can boost immunity are also available.
In short, barriers associated with conventional methods were largely resolved by the application of genomics technology, which greatly aids the development of innovative vaccines. However, arduous validation and the need for animal testing and clinical trials are still major technical issues that delay vaccine production. Another issue is the limited availability of online databases containing epitope data. To enhance the reliability of the genomics approach, structural biology data, immunogenicity data, and in silico B cell/T cell epitope data are critically required. The rapid evolution and advancement of the genomics approach brought on by structural and synthetic genomics is beyond expectations. Synthetic and structural genomics are crucial tools for future vaccine development as they can address MDR issues involved in the rise in intracellular bacterial pathogens. We are hopeful that further improvements in genomic approaches will lead to advances in the field, ultimately leading to improvements in existing vaccines and the discovery of new candidates.

 ,
, 


{kind=link}
{kind=link}
{kind=link}


