3.2. Supported Metal Oxide Catalysts
Transition-metal (e.g., Mn, Co, Cr, Ni, Fe, V, and Cu) oxides are considered to be practical and versatile catalysts for methane combustion and possess high catalytic activities owing to their multiple oxidation states, lattice defects (i.e., oxygen vacancies), strong oxygen mobility, easy desorption and activation of reactant molecules, and improved thermal stability. The low cost and facile synthesis of transition-metal oxide catalysts make them a prominent research topic, and much attention has been paid to the oxide structures accountable for the good performance. The cation states (e.g., Co
3+/Co
2+, Sn
4+/Sn
2+, Fe
2+/Fe
2+, Cu
2+/Cu
+/Cu
0, and Mn
4+/Mn
3+/Mn
2+) in the crystal structures are the most likely active sites for adsorption and activation of reactants and O
2 as well as for the high catalytic performance. For instance, a high-throughput investigation carried out by Saalfrank and Maier in 2004 substantiated the good performance of the noble-metal-free catalysts for CO oxidation [
67].
The transition metal (e.g., Cu, Fe, Co, Ni, Mn, and Ce) oxides with typical multiple valence states help to form redox cycles of the catalytic process between the high and low oxidation states, thereby restoring and releasing the lattice oxygen species. Phase transition of the active components may affect thermal stability and catalytic performance of the catalyst [
68]. Co
3O
4 has the weakest metal–oxygen bond, and the easy reduction of Co
3+ in Co
3O
4 to Co
2+ could accelerate formation of the oxygen vacancies at low temperatures [
69]. CeO
2 is one of the most effective metal oxides due to its excellent redox property and oxygen storage ability [
70]. However, the high-temperature redox cycle presents a remarkable challenge for structure and reactivity of the catalyst and limits the oxygen-carrying capacity of pure CeO
2. MnO
x has flexible valence states (i.e., Mn
2+, Mn
3+, and Mn
4+), among which the nanocubic MnO
2 sample exhibited the best low-temperature reducibility [
71]. ZrO
2 as an excellent stabilizer can effectively overcome the sintering challenge caused by a sharp decrease of surface area in the high-temperature redox cycle [
72]. However, thermal stability of the unmodified active NiO catalysts was poor, which could be combined with other metals for the utilization of catalytic lean methane combustion [
73]. For methane combustion in the presence of sulfur dioxide, metal oxide catalysts can form sulfates at a high temperature (e.g., 550 °C), which gives rise to an irreversible deactivation of the catalysts.
Most of the transition metals exhibit a virtually constant heat of adsorption in a large range of oxygen coverage; however, the chemisorption heat produced on noble metals (Rh, Pd, and Pt) decreases continuously with the rise in oxygen coverage [
74]. Cobalt oxides are one of the most widely investigated transition-metal oxides. Co
3O
4 is the most stable oxide in the range of 350–900 °C, and shows the best catalytic activity due to its two valence states (Co
3+ and Co
2+). Above 900 °C, Co
3O
4 loses oxygen spontaneously to form CoO. Co
3O
4 displays an ideal spinel-type structure, in which one-eighth of the tetrahedral sites is occupied by Co
2+ ions, whereas half of the octahedral sites is occupied by Co
3+ ions. The copresence of Co
3+ and Co
2+ pairs in the same catalysts seems to be critical for the good performance. The Co
3O
4/CeO
2 catalyst was used to study methane combustion under the reaction conditions of (1.0 vol% CH
4 + 4.0 vol% O
2 + Ar (balance) and SV = 30,000 mL/(g h)) [
75]. The best CH
4 oxidation activity (
T50% = 401 °C and
T90% = 490 °C) was achieved over the sample with a Co/(Co + Ce) atomic ratio of 0.75, which was due to the enhanced oxygen vacancy concentration, good reducibility, and strong mutual interaction between Ce and Co. The supported three-component (12 wt% Co
3O
4 + 3 wt% Fe
2O
3 + 3 wt% MnO
2) monolithic catalysts on a mixture of modified Ce–Zr–O solid solution and YSZ–Al
2O
3 showed a superior performance (
T50% = 358 °C and
T90% = 378 °C at SV = 12,000 mL/(g h)), which was associated with the synergistic effect of cobalt, iron, and manganese oxides and the high thermal stability for methane combustion [
76].
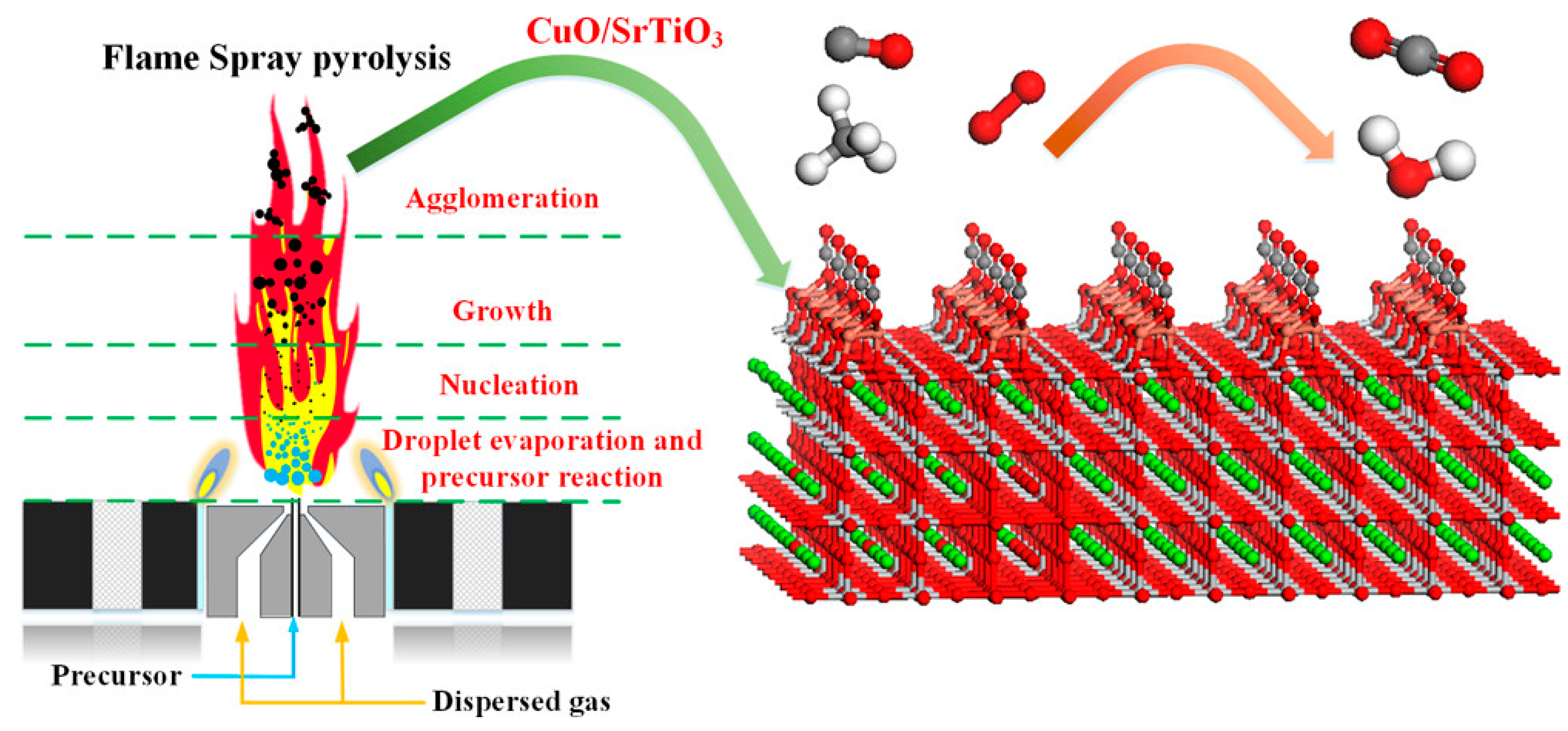
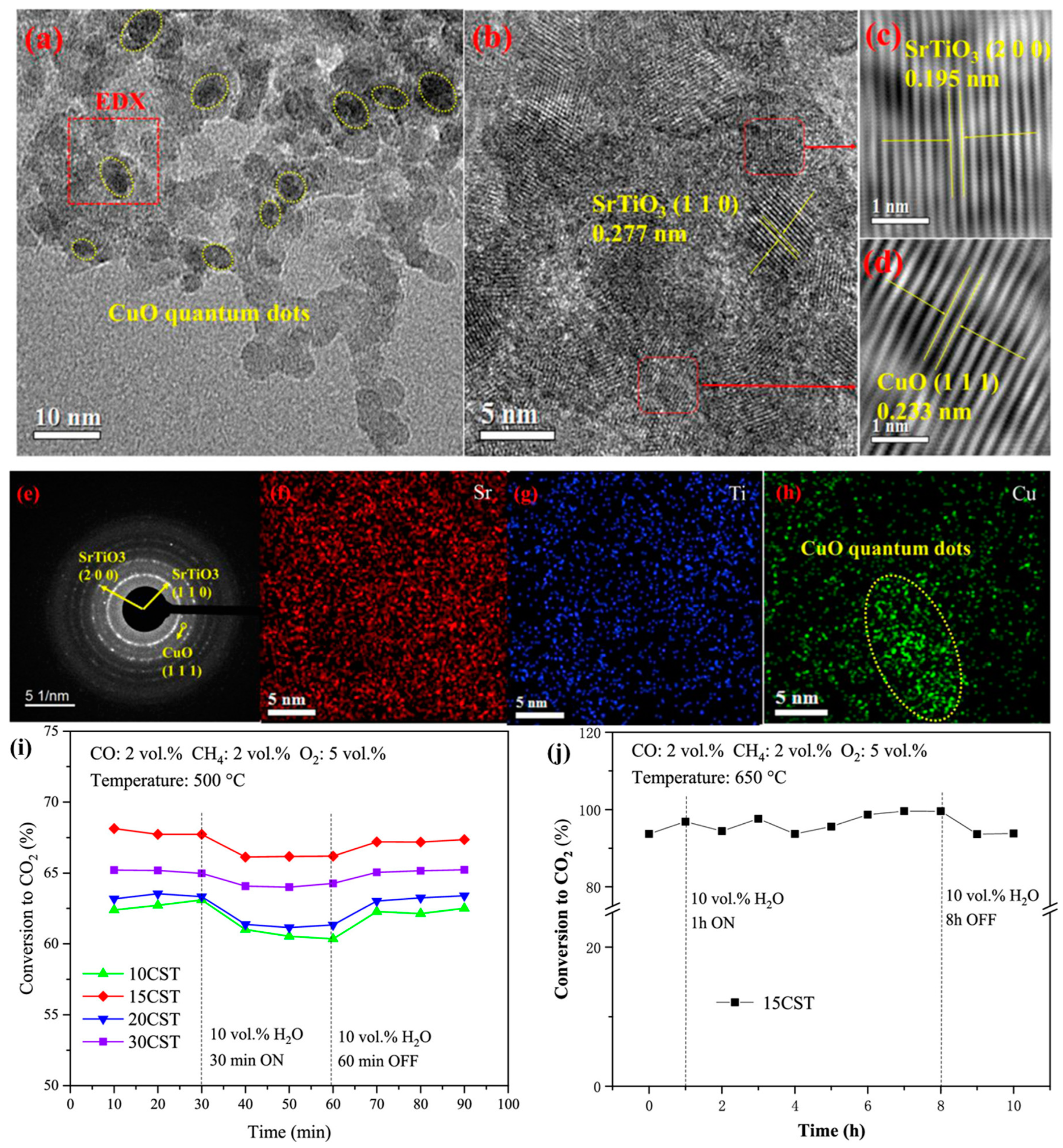
The Al
2O
3- or ZrO
2-supported CuO, Co
3O
4, and NiO catalysts exhibited good activities for CO oxidation, which was associated with uniform dispersion of the active phases (copper, cobalt, and nickel oxides) on alumina or zirconia. The intrinsic activity was not enhanced over the supported Cr
2O
3 samples, which was due to poor dispersion of chromia on alumina or zirconia at high temperatures. The CuO/SrTiO
3 sample (
Figure 7 and
Figure 8) was synthesized by the one-step method, and used for methane combustion due to its low price, outstanding thermal stability, and extraordinary adaptability [
77]. The 15 wt% CuO/SrTiO
3 catalyst with a strong metal–support interaction and excellent low-temperature reducibility exhibited the optimal activity (
T50% = 527 °C and
T90% = 645 °C at SV = 60,000 mL/(g h)) and the highest TOF
Cu at 400 °C (1.08 × 10
−3 s
−1). The authors concluded that the SrTiO
3 perovskite could effectively weaken the copper sintering at high temperatures, which was beneficial for the resistance to sintering and water-induced deactivation. The TiO
2-supported CuO materials with different crystalline structures were prepared to investigate the support effect and catalytic activity for methane combustion [
78]. The 7 wt% CuO/rutile sample obtained a 90% methane conversion at 550 °C at SV = 30,000 mL/(g h). The authors claimed that the superior activity was related to the easily formed Cu species and various chemical states of Cu elements on the stable rutile crystallite. The SnO
2/In
2O
3 (i.e., 20 wt% tin oxide + 80 wt% indium oxide) sample showed a complete methane conversion at 600 °C and SV = 30,000 mL/(g h) [
79]. Deactivation of the SnO
2/In
2O
3 catalyst in the presence of a large amount of water vapor was irreversible. However, the as-prepared sample showed an enhanced SO
2-tolerant performance by loading of the highly SO
2-resistant Sn on In
2O
3. The authors found that the high performance was attributed to the extra crystal defection and enhanced oxygen vacancies due to the loading of SnO
2.
Manganese is usually used with other elements (e.g., Cu, Co, Ni, and La) to form manganites or manganates, which perform well due to the fact that manganese oxidation states are constantly oscillated between Mn
4+ and Mn
3+. For example, the manganese-cobalt oxide with a Co/Mn molar ratio of 5:1 achieved the highest methane conversion of 90% at 320 °C and SV = 36,000 mL/(g h) [
9], in which the authors pointed out that crystal defects in spinel-type Co
3O
4 were enhanced after do** an appropriate amount of manganese, which increased the number of the octahedrally coordinated Co
2+ ions, facilitated oxygen mobility, and accelerated the dihydroxylation process.
It is generally known that particle size, surface area, oxygen deficiency density, and porosity are important factors in governing catalytic performance of a catalyst; however, the major drawback of a bulk catalyst is its rapid deactivation caused by sintering of the metal active sites and deposition of carbon. A large surface area is beneficial for the good dispersion of the active sites on the support, and a uniform pore-size distribution is favorable for the inhibition of agglomeration of the active phases, hence enhancing the high-temperature stability and catalytic performance. Ceria is a widely used support for methane combustion, which exhibits a moderate activity (a low light-off temperature around 300 °C) and a large oxygen-storage capacity. Zeng et al. [
80] reported that the CeO
2/Co
3O
4 catalyst with double pore distributions provided a larger number of the active sites as well as the good gas circulation channels, which reduced the internal diffusion resistance and improved the catalytic performance for CH
4 reforming. Andrey et al. [
81] adopted a reverse microemulsion route to obtain ultrahigh dispersion of ceria on the discrete barium hexaaluminate that showed a good high-temperature industrial application potential. The 25 wt% CeO
2/BaAl
12O
19 sample exhibited a low light-off temperature of 400 °C with an
Ea of 145 kJ/mol. Complete methane conversion was achieved over 25 wt% CeO
2/BaAl
12O
19 at 600 °C and SV = 60,000 mL/(g h). However, full methane conversion performance was observed during the heating–cooling–restarting cycles in the presence of H
2S and H
2O at 1100 °C. The authors concluded that the superior methane oxidation activity of 25 wt% CeO
2/BaAl
12O
19 was related to its high surface area, low-temperature reducibility, high-temperature thermal stability, excellent poisoning resistance, and synergistic chemical/electronic effect.
Provided that the composite metal oxides do not react with each other under the synthesis conditions, the nanocomposite design and ultrahigh component dispersion achieved through the flexible facile route can be widely used in develo** the complex-oxide catalysts with good reactivity and thermal stability for methane conversion. Mesoporous nanocrystalline MgO/Al
2O
3 materials with surface areas of 253–302 m
2/g and an average pore size of 7.62 nm were generated using a simple sol–gel approach [
82]. The MgO/Al
2O
3 catalyst showed high methane oxidation activity as well as good resistance against carbon formation. The meso-Fe
2O
3-loaded ZrO
2 porous nanobelts with an Fe/Zr molar ratio of 0.25 displayed good methane oxidation activity (
T50% = 580 °C and
T90% = 650 °C at SV = 25,000 mL/(g h); and
Ea = 97.9 kJ/mol) [
63]. The mesoporous alumina-supported PdO sample performed well with a 90% methane conversion at 400 °C, which was related to the highly dispersed PdO NPs in the meso-Al
2O
3 channels [
83]. Zhang et al. generated a series of the cordierite monolith-based catalysts by adding CeO
2, ZrO
2, La
2O
3, or CeO
2–ZrO
2 as the promoter to form Cu–Mn-complex oxides and tested their methane combustion activities [
84]. The introduction of the promoter increased the surface area and appropriately adjusted the porous structure, which favored the ultrahigh dispersion of the active component and the enhancement of oxygen species concentration. The Cu–Mn–Zr–O/Al
2O
3/COR monolithic material showed a 90% methane conversion at 570 °C and SV = 20,000 mL/(g h).
Chen and coworkers [
85,
86,
87,
88,
89] generated the multicomponent catalysts by do** the oxygen-storing rare-earth elements, which exhibited enhanced hydrothermal stability and good CH
4 oxidation activities. This research group demonstrated that the addition of a rare-earth element to the catalyst improved H
2O tolerance significantly, and initial specific surface area was kept after annealing at 1000 °C. The ZnZrAlO
x composite with 15 wt% ZnO displayed the light-off and complete conversion temperatures for methane combustion of 278 and 314 °C at SV = 50,000 mL/(g h), while those in the presence of H
2O were 342 and 371 °C [
88], respectively. The TiZrAlO
x composite with 5 wt% TiO
2 performed the best for CH
4 oxidation, with the
T50% and
T90% being 254 and 280 °C at SV = 30,000 mL/(g h) [
89], respectively. Du et al. synthesized the cobalt-based monolithic catalysts supported on YSZ–
γ-Al
2O
3 or CeO
2–Y
2O
3–ZrO
2 using the impregnation method, and studied their catalytic activities for CH
4 oxidation [
85]. It was found that the CoAl
2O
4 phase was not formed in the mixture support, thus making the CeO
2–Y
2O
3–ZrO
2 or YSZ–
γ-Al
2O
3 (
γ-Al
2O
3 modified by Y
2O
3 and ZrO
2) a multifunctional catalytic support. The loading of 20 wt% Co
3O
4 on the support made the catalyst perform the best with a 90% methane conversion at 436 °C, which was related to its high surface area, rich oxygen vacancy density, and good thermal stability. Svensson et al. prepared the MgO-based nanocomposite catalysts by adding 20 wt% LaMnO
3 for methane combustion [
90]. The sample fabricated by surface precipitation of LaMnO
3 with the Mg nitrate precursor performed the best below 1100 °C. It was reported that NiO/Ce
0.75Zr
0.25O
2 with a surface area of 77.9 m
2/g displayed the
T50% and
T90% of 500 and 560 °C for CH
4 oxidation at SV = 36,000 mL/(g h) [
91], respectively. The reaction order determined was in good agreement with a pseudo-first order toward CH
4 concentration, and the rate-determining step was the interaction of the dissociatively O
ads species with CH
4; furthermore the
Ea values over NiO and Ce
0.75Zr
0.25O
2 were 63 and 74 kJ/mol, respectively. Chen et al. synthesized the CeO
2–Al
2O
3–ZrO
2-supported Fe
2O
3 catalysts using an impregnation strategy and tested their catalytic activities for methane oxidation [
86]. The 8 wt% Fe
2O
3-loaded sample performed the best with a 90% methane conversion at 530 °C and SV = 15,000 mL/(g h). The authors believed that the high performance of the 8 wt% Fe
2O
3-loaded sample was related to its good thermal stability, high surface area, and ultrahigh component dispersion. Zhang et al. generated the cubically-structured, cauliflower-shaped Ce
0.6Zr
0.35Y
0.05O
2-supported PdO catalyst with a high surface area of 129 m
2/g using a hexadecyl trimethyl ammonium bromide template [
92]. The 4 wt% PdO/Ce
0.6Zr
0.35Y
0.05O
2 sample with a stable active phase showed a higher methane conversion (
T50% = 320 °C and
T90% = 360 °C at SV = 50,000 mL/(g h)). The hysteresis phenomenon of methane conversion versus temperature was remarkably decreased by loading PdO. Dai and coworkers [
93] reported the 3D wormhole-like mesopore-structured cubic Ce
0.6Zr
0.35Y
0.05O
2-supported Ag
2O catalysts, in which 2.0 wt% Ag
2O/Ce
0.6Zr
0.35Y
0.05O
2 with a high surface area of 65 m
2/g showed the highest activity for methane oxidation (
T50% = 540 °C and
T90% = 600 °C at SV = 50,000 mL/(g h)), which was related to the stable active Ag
2O phase, strong oxygen storage-release ability, and large surface area. Chen et al. prepared the oxygen-storage Ce
0.35Zr
0.55Y
0.07La
0.03O
1.95- and La–Al
2O
3-supported Fe
2O
3 catalysts, and evaluated their methane combustion activities [
87]. The former sample presented a surface area of 83 m
2/g, over which the
T50% and
T90% for methane conversion were 516 and 565 °C at SV = 60,000 mL/(g h), respectively. The authors thought that the excellent catalytic activity was attributed to the large surface area, rich O
ads species, and strong interaction between Fe
2O
3 and Ce
0.35Zr
0.55Y
0.07La
0.03O
1.95.
Table 2 summarizes catalytic activities of the ordered porous composite oxide and supported metal oxide catalysts for methane oxidation reported in the literature. It can be realized from the above investigations that high methane combustion performance is associated with the unique well-ordered 3D pore structure, high surface area, good thermal stability, and abundant oxygen vacancies of a catalyst.
3.3. Supported Noble Metal Catalysts
Noble metals catalysts are studied for low light-off temperatures and good C–H activation ability. However, uniformly dispersed noble metals NPs are easily sintered and aggregated to form nanoclusters at high temperatures with increased sizes, leading to poor thermal stability and irreversible catalyst deactivation. The design and preparation of the active components with specific structures can greatly improve thermal stability of the catalysts, and the addition of a base metal to noble metal(s) presents an optimized noble metals utilization efficiency with a less cost. The noble metal reduction is one of the most promising prospects for commercial applications by enhancing the efficiency of noble metal utilization, such as monoatomic, noble metal transition metal alloy, core–shell structure design, and suitable support selection. Recently, Dai’s group successfully embedded partially Pt NPs in the skeleton of a 3DOM Mn
2O
3 support, which exhibited an excellent thermal stability and catalytic performance compared with the catalyst derived from the PVA-protecting reduction approach [
52]. Another innovative strategy was developed by coating noble metals with metal oxides to construct the shell (metal oxide)-core (noble metal) structure to protect the active component. **, and the Pt–Pd oxide (i.e., PdO–PtO
2) exhibited better catalytic activity than the metallic Pt
0–Pd
0 with a good thermal stability. The meso-Pd, meso-Pt, and meso-Pd
2.41Pt showed the
T50% and
T90% values of 350, 372, 303, and 368, 506, and 322 °C at SV = 100,000 mL/(g h), respectively, among which meso-Pd
2.41Pt performed the best, with the highest TOF
Pd+Pt and specific reaction rate of 0.59 × 10
−3 s
−1 and 4.46 μmol/(g
cat s) at 280 °C, respectively. Partial deactivation of the Pd
2.41Pt catalyst caused by the introduction of carbon dioxide or water vapor was reversible, while the deactivation caused by the addition of SO
2 was irreversible. The authors believed that the well-ordered and developed mesoporous structure and good oxygen activation ability were accountable for the high catalytic activity. Cargnello et al. [
98] developed a supramolecular approach to deposit a palladium (Pd) core and a ceria (CeO
2) shell on a modified hydrophobic alumina. The commercial alumina substrate was highly hydrophilic, while the active comment Pd@CeO
2 was hydrophobic. The hydrophobic Pd@CeO
2 structure presented an agglomeration instead of adhering to alumina, resulting in irreversible deactivation of such a catalyst. The authors aimed to develop an efficiency strategy to regulate special properties of alumina surface to be hydrophobic and deposit the active comment as a single unit on the substrate surface. The alumina surface was decorated by organosilane triethoxy(octyl)silane to ensure the surface would be covered by the formed alkyl chain, leading to a larger adsorption capacity of the Pd@CeO
2 structure without deactivation. The droplets of water deposited on the hydrophobic alumina were repulsed instead of the favorable interaction with the OH groups in alumina, which further confirmed the adopted tactic. The as-prepared sample showed a remarkable higher methane oxidation activity (
T50% = 250 °C
T90% = 300 °C at SV = 200,000 mL/(g h)), with the highest TOF and the lowest
Ea of 47 × 10
−3 s
−1 and 103 kJ/mol at 250 °C, respectively. Furthermore, the supported catalyst remained intact without segregation or agglomeration during the calcination process at 850 °C. The authors pointed out that excellent complete methane oxidation activity was associated with the strong metal–support synergy interaction, well-defined ordered mesoporous structure, and good thermal stability under the adopted reaction conditions.
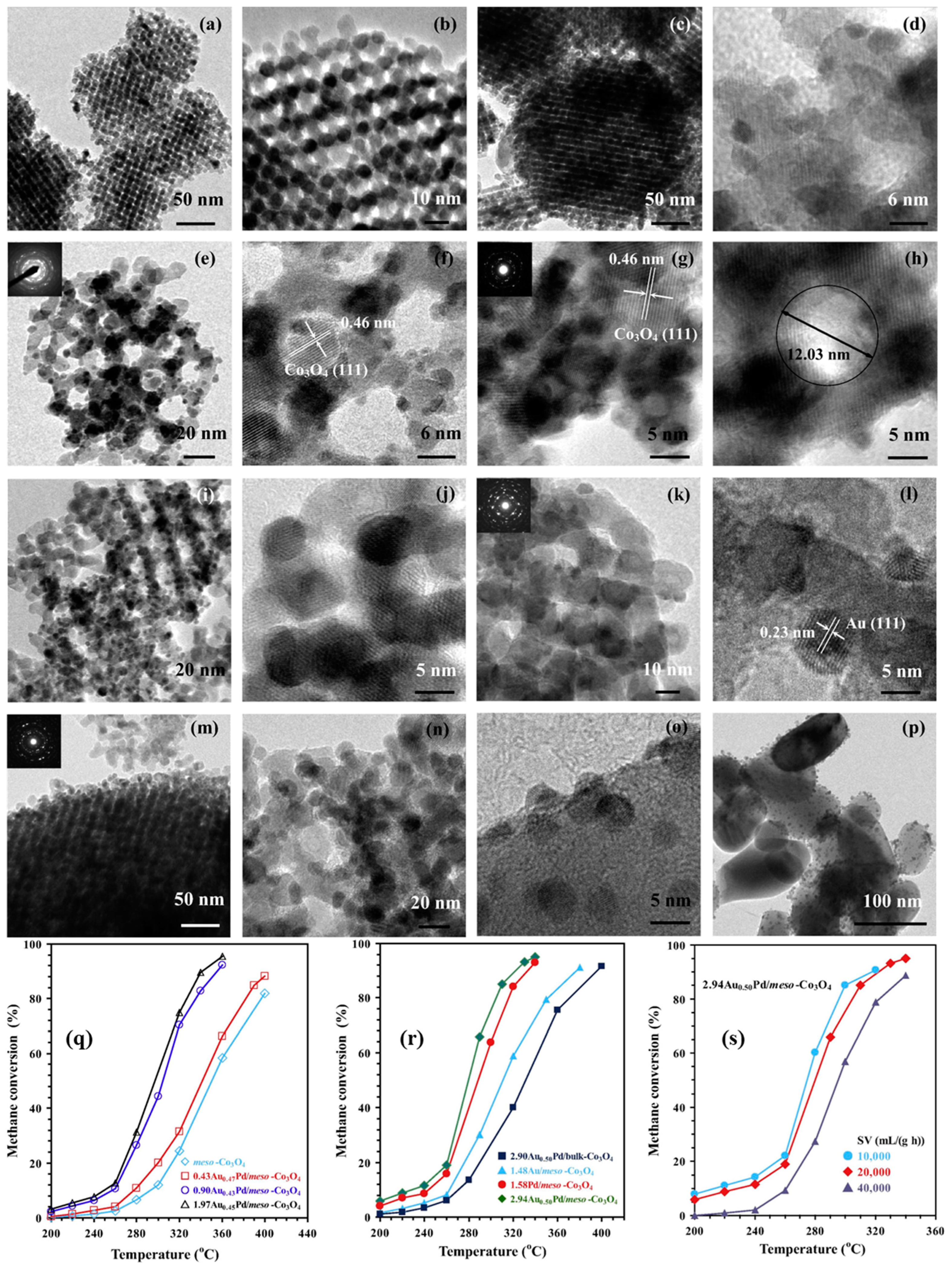
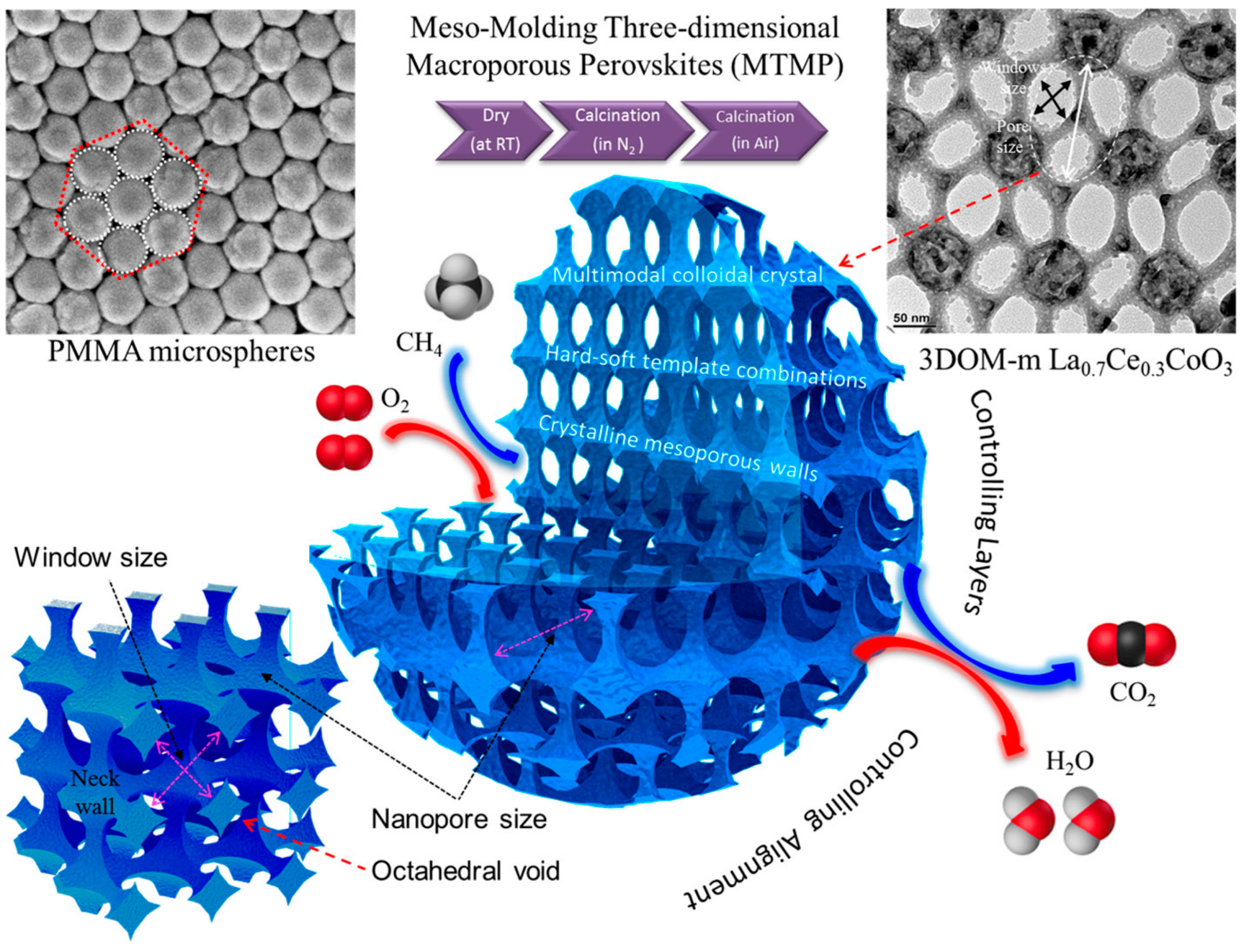
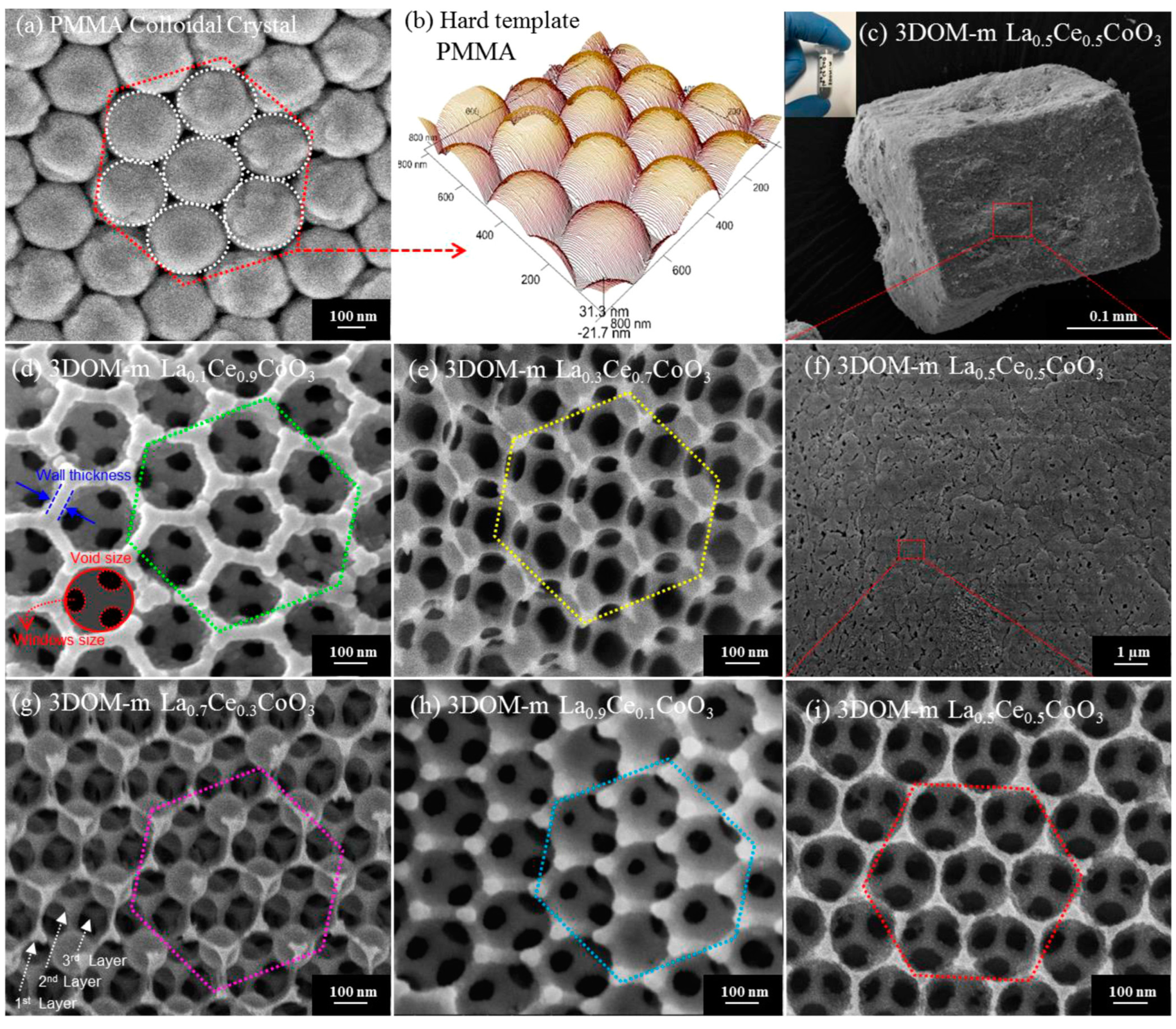
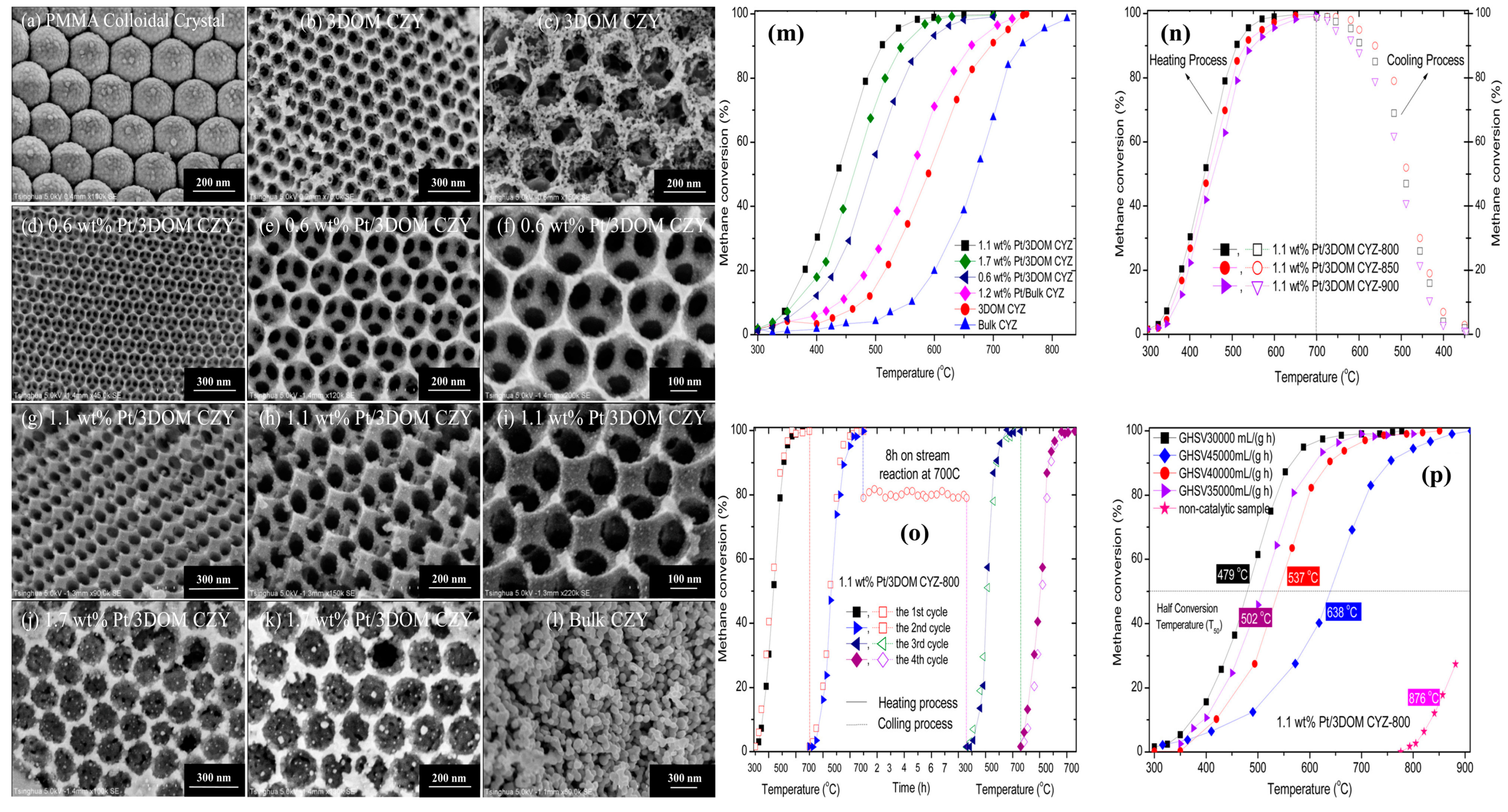
Generally speaking, it is accepted that the microstructure and function of single-noble-metal NPs can be modified by introducing a trace amount of a transition metal (Cr, Mn, Fe, or Co) or a noble metal (Au, Pd, Pt, or Ru). On this basis, Dai’s group investigated methane combustion over the supported noble–M (M = Au, Pd, Pt, Ru, Cr, Mn, Fe, and Co) NPs with enhanced oxygen activation ability and less noble metal use. Numerous experiments demonstrated that the facilely formed noble alloy NPs showed an excellent methane oxidation activity and good thermal stability as well as the valid noble metal utilization. Over the last 15 years, Dai’s group fabricated a series of the 3DOM-structured macro- or mesoporous mixed-metal-oxides-supported precious metal (Au, Pd, Pt, Ru, and alloy) catalysts with high surface areas and ordered porous channels using the PMMA- or KIT-6-templating and PVA-protecting reduction approaches, such as AuPd/meso-Co
3O
4 (mesopore diameter = 11–15 nm and surface area = 104–115 m
2/g) [
28], PdPt/meso-Mn
2O
3 (mesopore diameter = 9.3–12.6 nm and surface area = 76.5–95.3 m
2/g), AuRu/meso-Mn
2O
3 (mesopore diameter = 9.9–20.2 nm and surface area = 98.8–104.8 m
2/g) [
54], AuPd/3DOM CoCr
2O
4 (macropore diameter = 120–180 nm and surface area = 33–36 m
2/g) [
48], AuPd/3DOM La
0.6Sr
0.4MnO
3 (macropore diameter = 126–150 nm and surface area = 32.0–33.6 m
2/g) [
49], PdPt/3DOM LaMn Al
11O
19 (macropore diameter = 97–113 nm and surface area = 26.3–29.0 m
2/g) [
20,
50], PdPt/MnO
x/3DOM CoFe
2O
4 (macropore diameter = 83–123 nm and surface area = 19–28 m
2/g) [
51], Au–Pd–Co/3DOM Mn
2O
3 (macropore diameter = 175–205 nm and surface area = 36.9–38.5 m
2/g) [
25], Au–Pd–CoO/3DOM Co
3O
4 (macropore diameter = 115–185 nm and surface area = 27–47 m
2/g) [
26], CoPd/3DOM CeO
2 (macropore diameter = 145–165 nm and surface area = 36.7–41.2 m
2/g) [
27], Ag/3DOM La
0.6Sr
0.4MnO
3 (macropore diameter = 101 nm and surface area = 41.5 m
2/g) [
47], and Pt/3DOM Ce
0.6Zr
0.3Y
0.1O
2 (macropore diameter = 121–147 nm and surface area = 84–95 m
2/g) (
Figure 9) [
99]. Dai and coworkers systematically studied the correlation between noble metal and several diverse supports in methane oxidation. It was observed that AuPd NPs were uniformly distributed on the surface of meso-Co
3O
4 with an average size of 2.7–4.5 nm [
28]. The 2.94Au
0.50Pd/meso-Co
3O
4 exhibited an excellent activity and possessed the highest specific reaction rate at 260 °C (1.47 μmol/(g
cat s), 91.7 μmol/(g
noble metal s) or 1.50 μmol/(g
Co3O4 s)). Over the 2.94Au0.50Pd/meso-Co
3O
4 sample at a SV of 20,000 mL/(g h), the
T10%,
T50%, and
T90% were 230, 280, and 324 °C with the lowest
Ea and the highest TOF
AuPd of 44.4 kJ/mol and 12.23 × 10
−3 s
−1, respectively. Similarly, among the meso-Mn
2O
3-supported PdPt alloy (2.1–2.8 nm in particle size) samples [
100], 1.41(Pd
5.1Pt)/meso-Mn
2O
3 showed the best methane oxidation performance: The
T10%,
T50%, and
T90% were 265, 345, and 425 °C, respectively; the highest TOF
PdPt was 1.02 × 10
−3 s
−1, and the best specific reaction rate was 6.39 μmol/(g
cat s) or 6.48 μmol/(g
Mn2O3 s) at 400 °C. In addition, the as-prepared sample displayed the increased endurance performance in the presence of SO
2, CO
2, or H
2O by introducing an appropriate amount of Pt. Moreover, AuRu/meso-Mn
2O
3 was generated to improve the reducibility, and the 0.97AuRu/meso-Mn
2O
3 sample showed the highest activity (
T50% = 470 °C and
T90% = 540 °C at SV = 20,000 mL/(g h)) [
54]. The same group investigated the Cr-based spinel-type-oxides-supported gold–palladium alloy materials with abundant O
ads concentrations and good reducibility for methane combustion. The 1.93AuPd
1.95/3DOM CoCr
2O
4 catalyst exhibited the highest methane conversion (
T50% = 353 °C and
T90% = 394 °C at SV = 20,000 mL/(g h)) with the superior TOF
AuPd (0.271 × 10
−3 s
−1) and the highest specific reaction rate (1.15 μmol/(g
cat s)) at 320 °C [
48]. The presence of SO
2 in the reaction system over the 0.97AuRu/meso-Mn
2O
3 sample caused a permanent loss in activity, while the partial deactivation due to moisture introduction was reversible. The AuPd/3DOM La
0.6Sr
0.4MnO
3 bimetallic catalysts (
Figure 10) were prepared with a good thermal stability and rich adsorbed oxygen species [
49], among which 2.92AuPd/3DOM La
0.6Sr
0.4MnO
3 with the appropriate Au/Pd molar ratio of 1:2 exhibited the
T50% and
T90% values of 314 and 336 °C at SV = 50,000 mL/(g h) for CH
4 oxidation. This sample exhibited the highest TOF
AuPd (10.2 × 10
−3 s
−1) and the lowest
Ea (72.4 kJ/mol). The size of the bimetallic Au–Pd catalyst increased from 2.15 to 3.76 nm upon exposure to water, indicating that the presence of water vapor caused an irreversible deactivation. AAl
12O
19 is regarded as a promising catalyst candidate due to its sintering temperature at above 1200 °C (i.e., it possesses a good thermal stability). The same research group also tested methane combustion activities over PdPt/3DOM LaMnAl
11O
19 with noble metal particle sizes of 3–5 nm [
20,
50]. The 1.14Pd
2.8Pt/3DOM LaMnAl
11O
19 sample showed the best activity, with the
T10%,
T50%, and
T90% values being 284, 372, and 456 °C at SV = 20,000 mL/(g h), respectively. The ex situ X-ray photoelectron spectroscopy (ex situ XPS) was used to investigate formation of the active PdO species during the oxidation process of Pd in 1.14Pd2.8Pt/3DOM LaMnAl
11O
19 at different temperatures. Do** with an appropriate amount of Pt to the supported Pd catalyst could improve H
2O, CO
2, and SO
2 tolerance. Recently, the same group investigated the ternary catalysts (which were combined with noble metal NPs, transition-metal oxide, and porous spinel-type mixed oxide) for methane combustion. The as-prepared PdPt/MnO
x/3DOM CoFe
2O
4 sample with a PdPt particle size of 2.2–3.0 nm exhibited the highest O
ads species concentration and the best low-temperature reducibility [
51]. The 1.81PdPt/6.7MnO
x/3DOM CoFe
2O
4 sample showed the highest activity (
T10% = 255 °C,
T50% = 301 °C, and
T90% = 372 °C at SV = 20,000 mL/(g h);
Ea = 59 kJ/mol; TOF
Noble metal = 37.44 × 10
−3 s
−1, and TOF
CoFe2O4 = 0.28 × 10
−3 s
−1).
After a brief discussion, we can realize from the data summarized in
Table 3 that methane combustion activity over the various 3DOM-structured mixed-metal-oxides-supported noble metal (Au, Pd, Pt, Ru, and alloy) catalysts decrease in the order of 2.94Au
0.50Pd/meso-Co
3O
4 (
T90% = 324 °C) > 2.92AuPd/3DOM La
0.6Sr
0.4MnO
3 (
T90% = 336 °C) > 1.81PdPt/6.7MnO
x/3DOM CoFe
2O
4 (
T90% = 372 °C) > 1.93AuPd
1.95/3DOM CoCr
2O
4 (
T90% = 394 °C) > 1.41(Pd
5.1Pt)/meso-Mn
2O
3 (
T90% = 425 °C) > 1.14Pd
2.8Pt/3DOM LaMnAl
11O
19 (
T90% = 456 °C) > 0.97AuRu/meso-Mn
2O
3 (
T90% = 540 °C). The authors assigned high methane oxidation activity of the typical catalyst to its high surface area with well-ordered and developed porous structure, abundant active oxygen species, good thermal stability, good low-temperature reducibility, and strong interaction between noble metal NPs and 3DOM-structued support.
Supported noble metals catalysts are generally divided into two forms: metallic state and oxidized state. The oxygen on the surface of the metal phase is usually adsorbed oxygen, which exhibits a low adsorption energy. Methane dissociation is generally considered to be a rate-determining step, and reaction kinetics are discussed according to the Eley-Rideal (E-R) [
101] and Langmuir-Hinshelwood (L-H) mechanisms [
102]. Metal oxides always coordinate with metal atoms and lattice oxygen, where the adsorption energy of lattice oxygen is higher than that of the adsorbed oxygen. The consumed lattice oxygen during the catalytic methane oxidation process is then made up by gaseous oxygen. In this case, the Mars-van Krevelen (MvK) mechanism is proposed to discuss the redox process [
103]. Methane and oxygen are adsorbed on the surface of a catalyst according to the L–H mechanism. Different intermediates are formed by adsorption and activation of methane during the dehydrogenation process. The dehydrogenation process from CH
4 to the C* species on the Pd surface was revealed by the first-principles simulation [
104]. The reaction process of the adsorbed methane and oxygen species can be described using the following steps [
105]:
CH4 + * → CH4*.
O2 + 2* → 2O*.
CH4* + O* → CH3* + OH*.
CH3* + O* → HCHO* + H*.
HCHO* + O* → CHO* + H*.
CH* + * → C* + OH*.
C* + O* → CO* + *.
CO* + O* → CO2* + *.
CO2* → CO2 + *.
OH* + OH* → O* + H2O*.
H2O* → H2O + *.
Here, * represents the active adsorption sites, and X* represents the adsorbed X species at the active sites.
The reaction process of the gaseous methane and adsorbed oxygen species in the E-R mechanism can be described using the following steps:
O2 + 2* → 2O*.
CH4 + 2O* → HCHO* + OH*.
HCHO* + O* → CHO* + OH*.
CHO* + O* → CO* + OH*.
CO* + O* → CO2* + *.
CO2* → CO2 + *.
OH* + OH* → * + H2O*.
H2O* → H2O + *.
The mechanism of methane oxidation with lattice oxygen vacancy formation via the MvK redox steps over PdO (101) was investigated. Rate-controlling analysis reveals that the dissociative CH
4 adsorption via hydrogen abstraction over the Pd
cus-O
cus site-pairs was the rate-determining step during the light-off process. Methane is first adsorbed on the surface of a catalyst and then reacts with lattice oxygen to form CO
2 and H
2O in the MvK redox mechanism. Detailed methane oxidation reaction path analysis (RPA) has been discussed for the dry and the wet reaction mixture with 12 vol% water in the feedstock at 1 bar to elucidate the preferential pathway. Methane is adsorbed and reacted with lattice oxygen to generate the side products in sequence under the dry condition and temperatures of 200–400 °C: CH
4 → CH
3 → CH
2OH → CH
2O → CHO → CO → CO
2. Molecular H
2O adsorption or desorption contributes to vacancy formation through O
2 adsorption from gas-phase and hydroxide formation or decomposition. The preferential path under the dry condition and temperatures above 400 °C is similar to that under the wet condition: CH
4 → CH
3 → CH
2 → CH
2O → CHO → CO → CO
2. The major difference is reaction sequence of the hydroxyl-methyl intermediates, which is due to water competitive adsorption [
106].
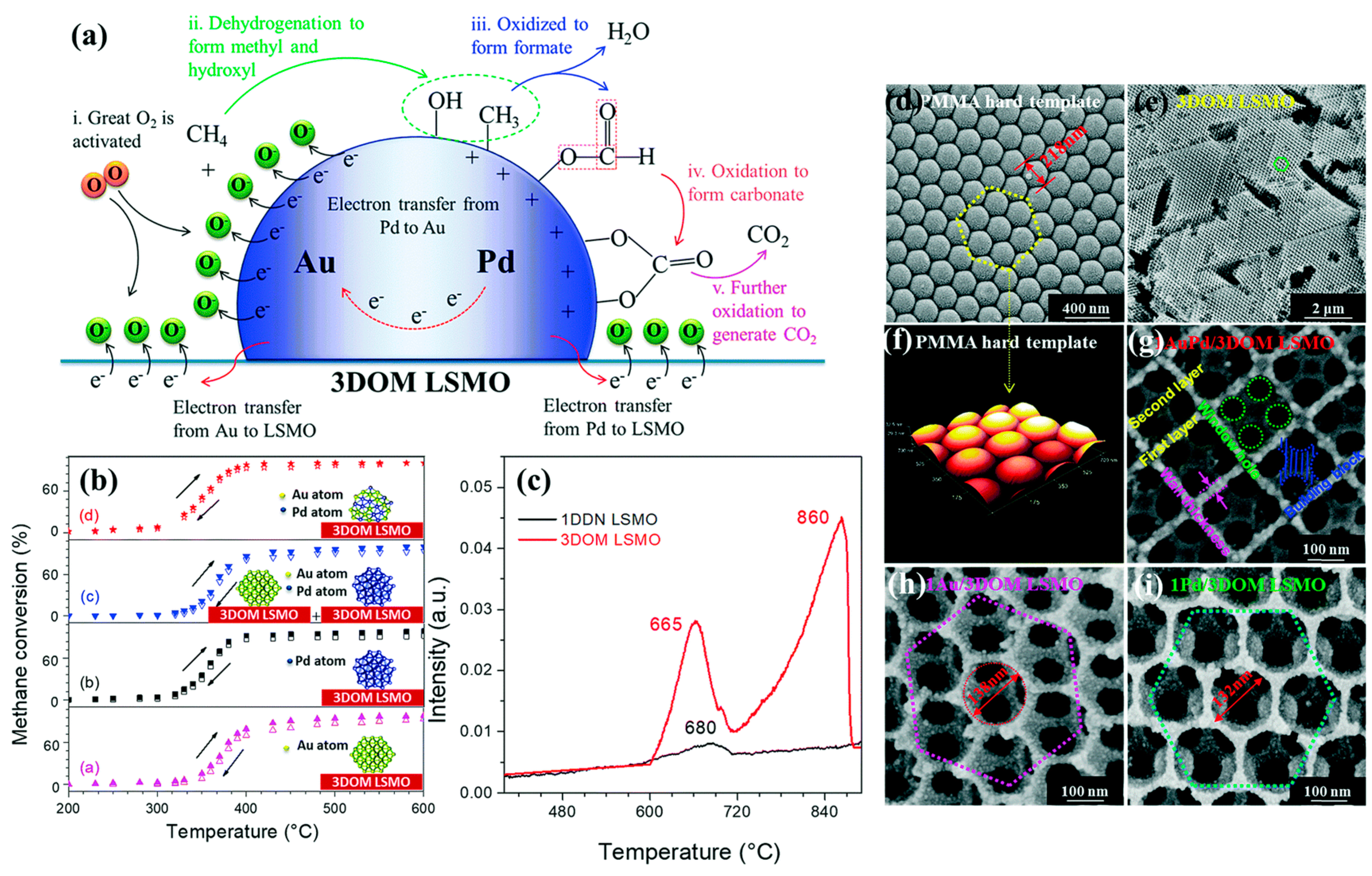
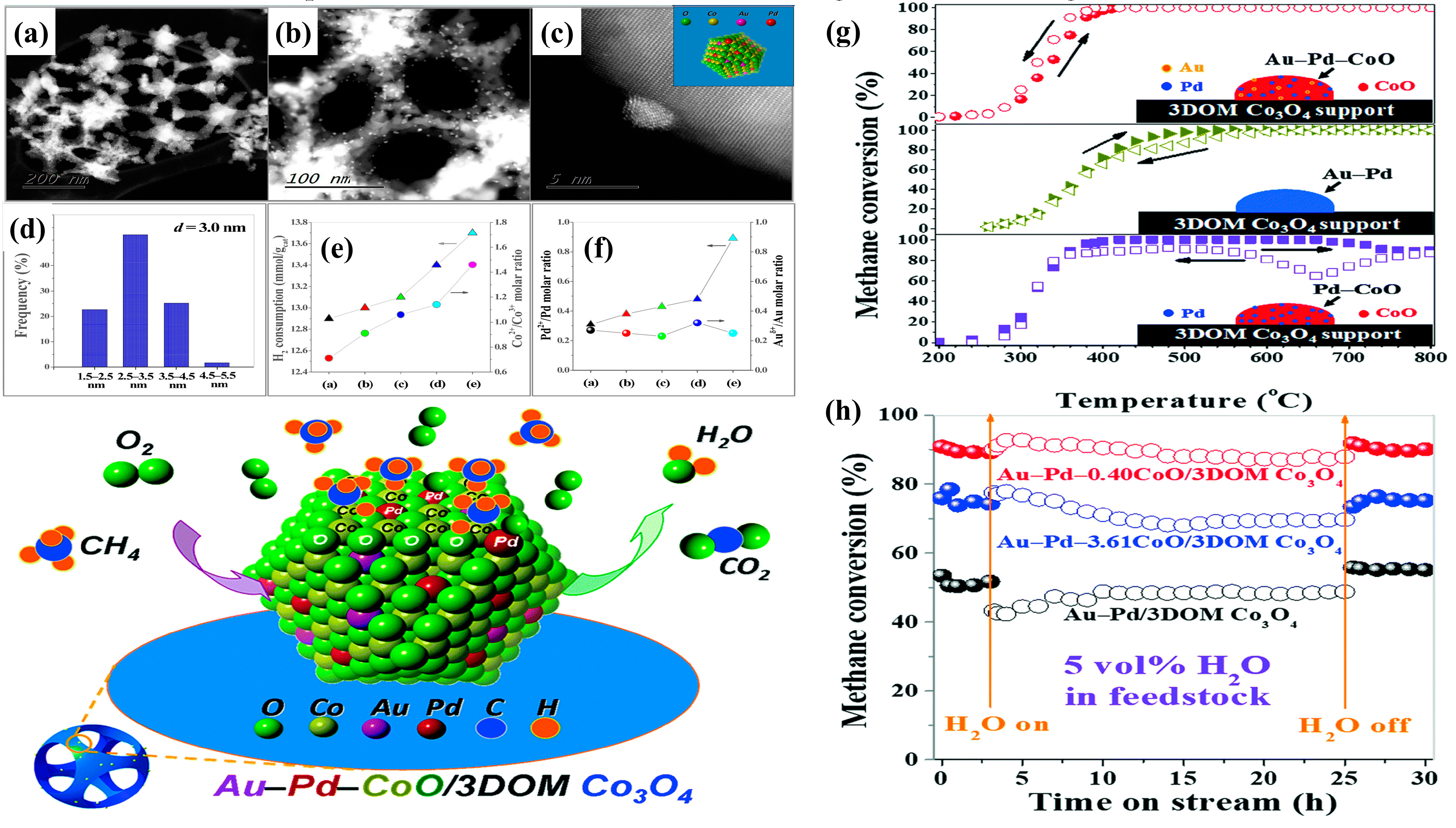
To probe the mechanism of CH
4 combustion over the 3DOM-structured metal-oxide-supported bimetallic NPs, methane temperature-programmed desorption (CH
4-TPD) and in situ reflectance Fourier transform infrared spectroscopic (in situ DRIFTS) experiments were intensively carried out by Dai’s group. The Au-Pd-
xCoO/3DOM Co
3O
4 sample (
Figure 11) retained an excellent sustainable catalytic performance by do** an appropriate amount of CoO [
26]. The cobalt was present in the form of a CoO phase, and the PdO-CoO interface was generated in the supported Au-Pd sample. Au-Pd-0.4CoO/3DOM Co
3O
4 showed the highest methane oxidation activity (
T50% = 312 °C and
T90% = 341 °C at SV = 20,000 mL/(g h)) with the highest TOF
Pd (11.8 × 10
−3 s
−1) and the highest specific reaction rate (110.5 μmol/(g
Pd s)) at 280 °C, which was a result due to formation of the unique PdO-CoO interface in the supported noble metal catalyst that was beneficial for CH
4 activation and O
ads species concentration enhancement. The CoO inclusion significantly favored the generation of Pd
2+ species and facilitated the inhibition of hydroxyl formation, greatly improving moisture tolerance. The CH
4-TPD profile displayed a lower methane desorption temperature (345 °C) on the CoO surface in Au–Pd–
xCoO/3DOM Co
3O
4, which could be owing to the strong interaction between PdO and CoO and enhanced O
ads species concentration. Methane could be activated flexibly on the PdO surface in Au–Pd–
xCoO/3DOM Co
3O
4, and the adsorbed methane reacted with the O
ads species to form CO
2 and H
2O. Methane oxidation species generated over the Au–Pd–3.61CoO/3DOM Co
3O
4 sample were detected by the in-situ DRIFTS experiments using an O
2/CH
4 (molar ratio = 4) gas mixture. The obtained intermediate products, such as the H
2O, CH
4, CO
2, CH
3−, H
2CO, HCOO
−, and CH
2OH
− species, further demonstrated that CoO was the active site for methane adsorption and activation. In addition, Co was doped into Pd/3DOM CeO
2 to generate a high O
ads or CH
4 concentration and display good thermal stability [
27]. Co was oxidized into CoO
x easily at 340–450 °C, which gave rise to the enhanced methane oxidation performance, and PdCo
x exhibited a better thermal stability than Pd or Co. The Co
3.5Pd/3DOM CeO
2 sample exhibited the best methane combustion activity (
T50% = 430 °C and
T90% = 480 °C at SV = 40,000 mL/(g h)) with the lowest
Ea (58 kJ/mol), the highest specific reaction rate (1334.3 × 10
−6 mol/(g
Pd s)), and the highest TOF
Co (0.04 s
−1).
Similarly, it was reported that the addition of gold to AuPd/3DOM La
0.6Sr
0.4MnO
3 accelerated the methane oxidation rate [
49]. The in-situ DRIFTS absorption bands of Pd/3DOM La
0.6Sr
0.4MnO
3 could be separated into three significant spectral zones, which led to a more efficient methane combustion mechanism to facilitate complete oxidation. The first-zone absorption bands were attributed to gas-phase CH
4 and CH
3−. The antisymmetric stretching vibration at 3014 cm
−1 and the deformation vibration at 1300 cm
−1 of the C–H bonds were ascribed to the CH
4 molecule, while the absorption band at 1450 cm
−1 belonged to the asymmetric stretching of C–H in CH
3−. The second-zone absorption bands were attributed to gas-phase CO
2 with the absorption bands at 2330 and 2360 cm
−1 due to the C=O stretching vibration. The third-zone absorption bands were related to the other byproduct species. The bands at 1060 and 1155 cm
−1 were ascribed to the O–O stretching vibration of the O
2− species, while the ones at 1250 and 1415 cm
−1 represented the stretching vibration of C–H in formaldehyde. The strong C–O asymmetric stretching vibration at 1550 and 1690 cm
−1 as well as the C=O stretching vibration at 1840 cm
−1 were associated with the formate species. A new bidentate carbonate species appeared at 1170 cm
−1 for the AuPd/3DOM La
0.6Sr
0.4MnO
3 catalyst, instead of stretching vibration (at 1250 and 1415 cm
−1) of C–H in formaldehyde (H
2CO) for the Pd/3DOM La
0.6Sr
0.4MnO
3 catalyst. The reaction rate was determined by the activation of C–H bond. The C–H bonds at 1300 cm
−1 appeared at 200 and 300 °C over AuPd (or Pd)/3DOM La
0.6Sr
0.4MnO
3, which revealed that the do** of Au gave rise to the lower CH
4 activation temperature and higher methane oxidation activity. The methane oxidation mechanism over AuPd/3DOM La
0.6Sr
0.4MnO
3 can be summarized as two separate sequences of elementary steps based on the in-situ DRIFTS results: The O
2 is activated by the involved transferred electrons to form O
ads species on the more electronegative surface caused by Au do**, thus the formed O
ads species cause the C–H bonds in CH
4 to dehydrogenate into CH
3− and OH
−. The CH
3− species is subsequently bonded to the unsaturated sites and dissociated into the HCOO
− species, or the CH
3− species is further H-abstracted directly into the H
+ and HCOO
− species (which explains the lack of the CH
2O species in the in-situ DRIFTS spectra over AuPd/3DOM La
0.6Sr
0.4MnO
3 compared with those over Pd/3DOM La
0.6Sr
0.4MnO
3). In addition, the above result indicated that the CH
3− species could transfer freely and interact with the additional O
ads species more readily, leading to acceleration of the oxidation reaction. The H
+ could combine with O
ads to generate H
2O, while H
2CO was first activated to form HCOO
− and then completely oxidized to CO
2 and H
2O.
3.4. Effects of H2O and SO2 on Methane Combustion
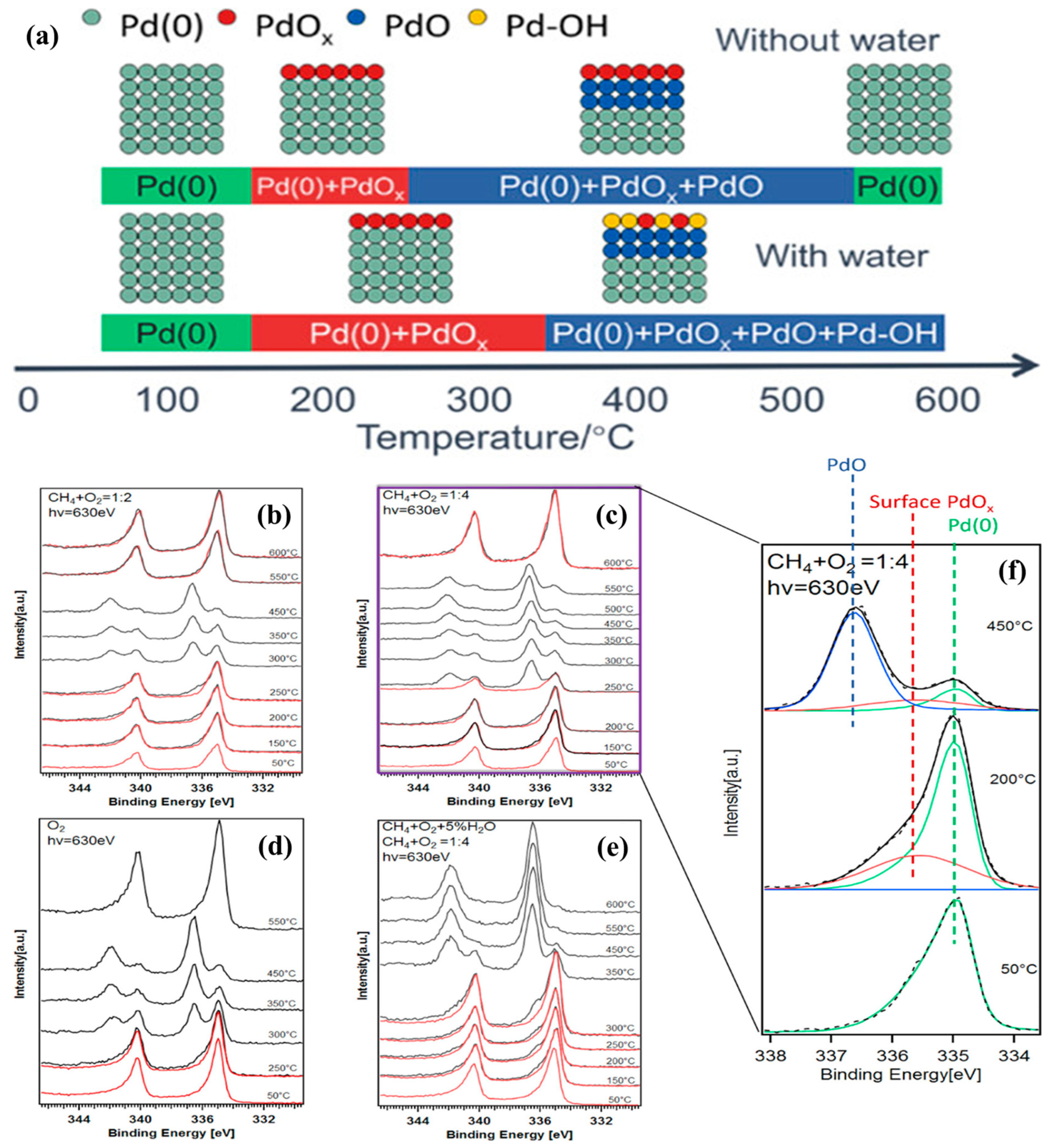
H
2O poisoning was a significant challenge for the supported noble metal materials in methane combustion at low temperatures. Bokhoven’s group investigated the electronic states of palladium (
Figure 12) and the effect of water codosing on CH
4 combustion using the ambient-pressure X-ray photoelectron spectroscopic (APXPS) technique [
107]. The evolution of metallic palladium (Pd
0 at binding energy (BE) = 334.95 eV), surface palladium oxide (PdO
x at BE = 335.6 eV), and bulk palladium oxide (PdO at BE = 336.7 eV) was studied in a gas mixture of CH
4/O
2 with different ratios and the water coexisting case at 50–600 °C. The Pd
0 species was detected in the initial stage at 50 °C. The fraction of the surface PdO
x species reached the maximal value at a certain temperature of 100–200 °C for the CH
4/O
2 mixture and prolonged to 300 °C both in the pure O
2 and H
2O cofeeding cases. The PdO species appeared in the absence of water at 250 °C, and was completely reduced to the Pd
0 species when the temperature reached 550 or 600 °C. However, the peak corresponding to the PdO species appeared in the presence of H
2O only at 350 °C, and no quantitative reduction of PdO
x happened, even at 600 °C. It was concluded that water inhibited the conversion of methane by converting the PdO
x species to the Pd–OH species. Palladium hydroxyls blocked the coordinatively unsaturated Pd sites (Pd
cus) for methane adsorption and the adjacent oxidized sites (Pd–O) for the activation of methane to intermediates, instead of forming hydroxyl groups. Water was adsorbed at the surface oxygen vacancies and deprotonated to form a second hydroxyl at a neighboring Pd–O site; thus, water accumulation dramatically hindered the adsorption and migration of O
2 to restrict regeneration of the active sites as well as to suppress formation of the active PdO phase.
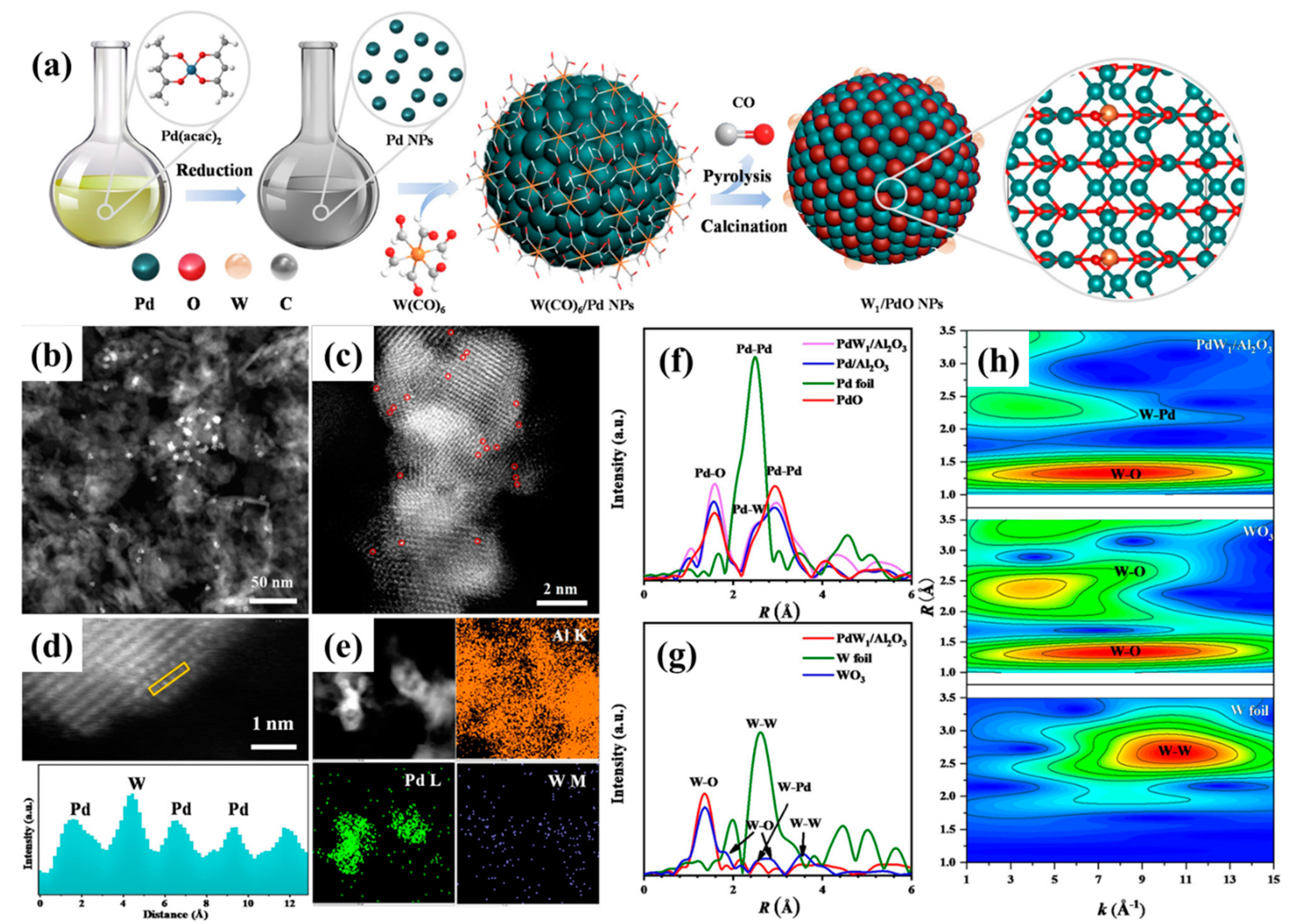
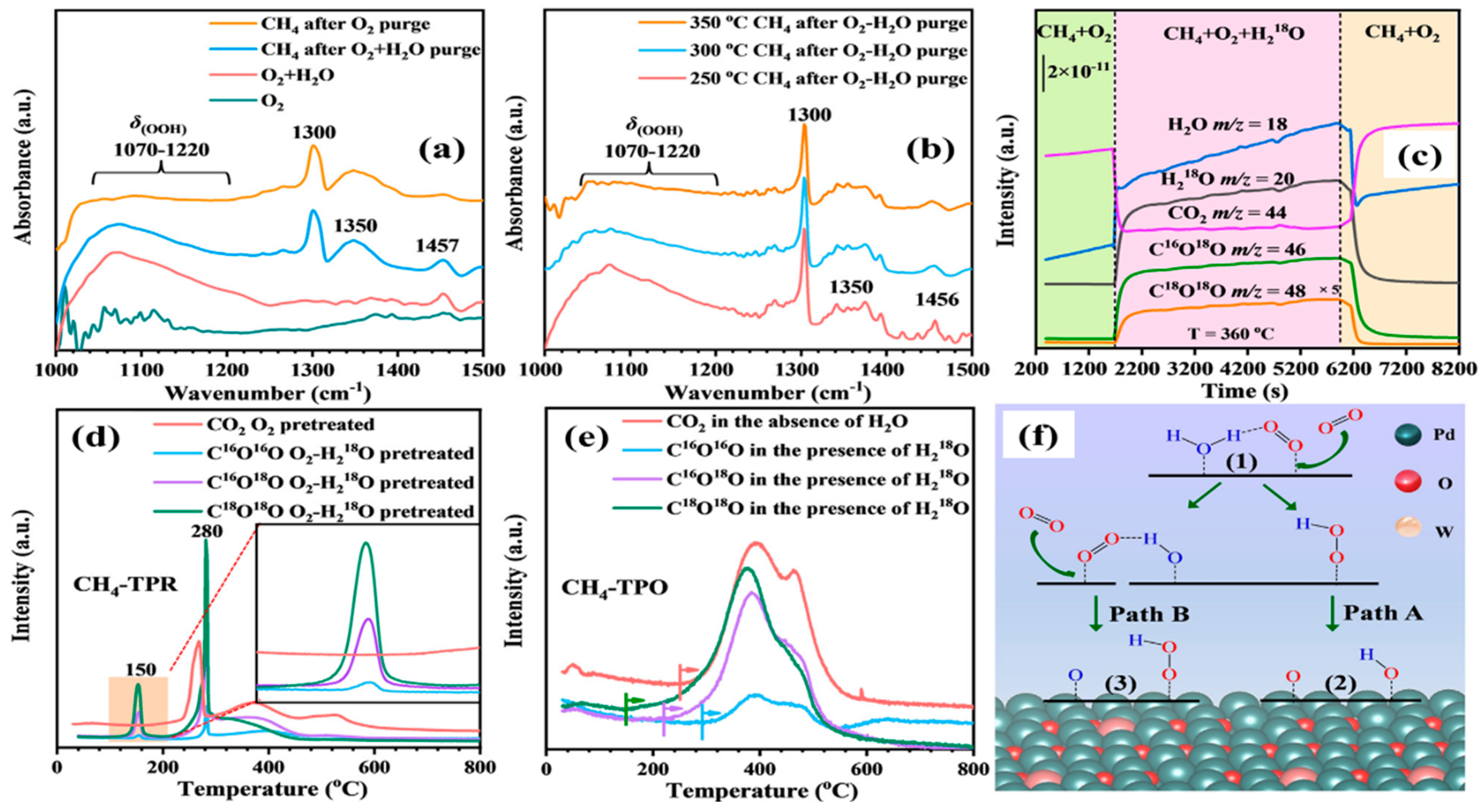
Dai and coworkers [
108] fabricated the water-resistant catalysts for CH
4 low-temperature combustion using the stepwise strategy to generate the atomically dispersed tungsten species with tailored local coordination structures and electronic states at the catalytically active sites on Pd NPs (
Figure 13 and
Figure 14). The Pd-O-W-like nanocomposite formed on the PdO surface with an atomic scale interface was responsible for the outstanding catalytic activity. The water adsorption energy decreased from 1.23 eV (at the Pd site on PdO) to 0.83 eV (at the Pd site on Pd-O-W), indicating a valid inhibition of water adsorption on the supported PdW sample, which effectively mitigated lattice oxygen deactivation obviously in the presence of H
2O. Moreover, the oxygen adsorption energy downgraded from 1.12 eV (at the Pd site on PdO) to 1.99 eV (at the Pd site on Pd-O-W), further confirming a larger oxygen adsorption capacity of the supported PdW catalyst for excellent catalytic performance. The supported catalyst preferred a better resistance to water poisoning than the conventional catalysts for CH
4 oxidation via a hydroperoxyl-promoted species and a novel coexistence reaction mechanism of Mars van–Krevelen and Langmuir–Hinshelwood models over the catalyst in the presence of moisture. DFT calculations revealed that the upshift of the
d-band center of palladium caused by the electron transfer from the atomically dispersed tungsten in the supported PdW sample was expected to enhance the oxygen adsorption and activation ability, which was associated with superior methane combustion activity and excellent water tolerance.
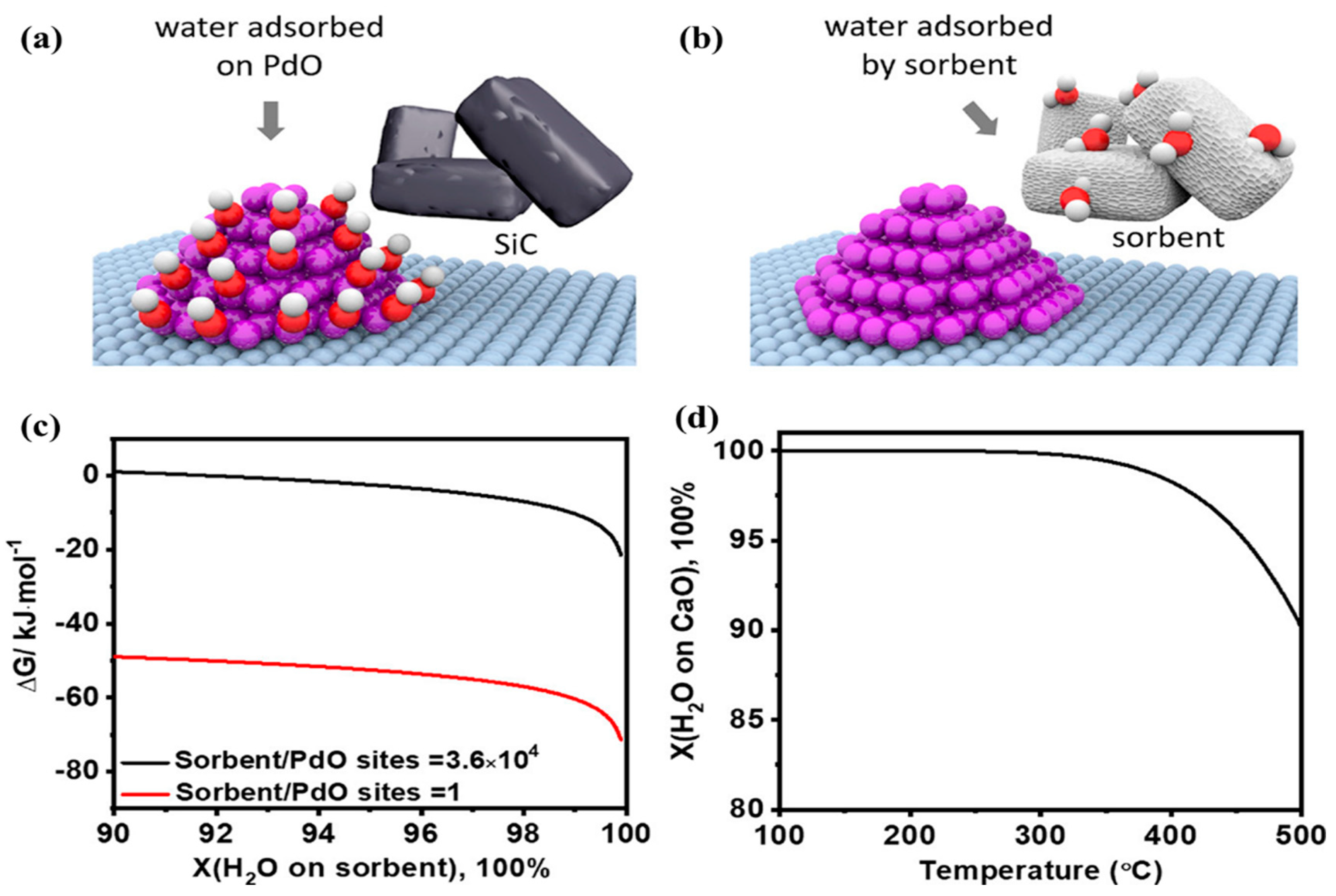
Cargnello and coworkers [
109] developed an in situ water sorption strategy to remove the water formed during the process of CH
4 oxidation using zeolite, alumina, or CaO as a water sorbent (
Figure 15). The do** of zeolite or Al
2O
3 to the supported Pd catalyst could transiently decrease H
2O concentration in a limited amount, thus leading to enhancement in activity below 300 °C. The hydroxylation of PdO happened quickly due to the limited number of sorption sites on zeolite or Al
2O
3, resulting in a poor activity of the Pd catalyst. On the contrary, the Pd catalyst diluted with a stronger CaO exhibited a sixfold improvement transient activity, which maintained a 10 h on-stream reaction compared with the sorbent-free sample. A thermodynamic model and detailed Gibbs free energy calculations were established to further prove the water durability of CaO. Although Gibbs free energy of water sorption on CaO was −21 kJ/mol with a sufficient number of sites, it was higher than that (−38.5 kJ/mol) of H
2O adsorption on PdO at 300 °C. The number of the adsorptive sites was equal to that on CaO, which was 3.6 × 10
4 times more than that of the PdO sites, and the complete water sorption at the CaO sites could still be achieved due to the large number of the adsorptive sites. Based on the above results, mechanistic analysis declaimed that the water sorption efficiency was influenced by both sorption energy and number of the adsorptive sites.
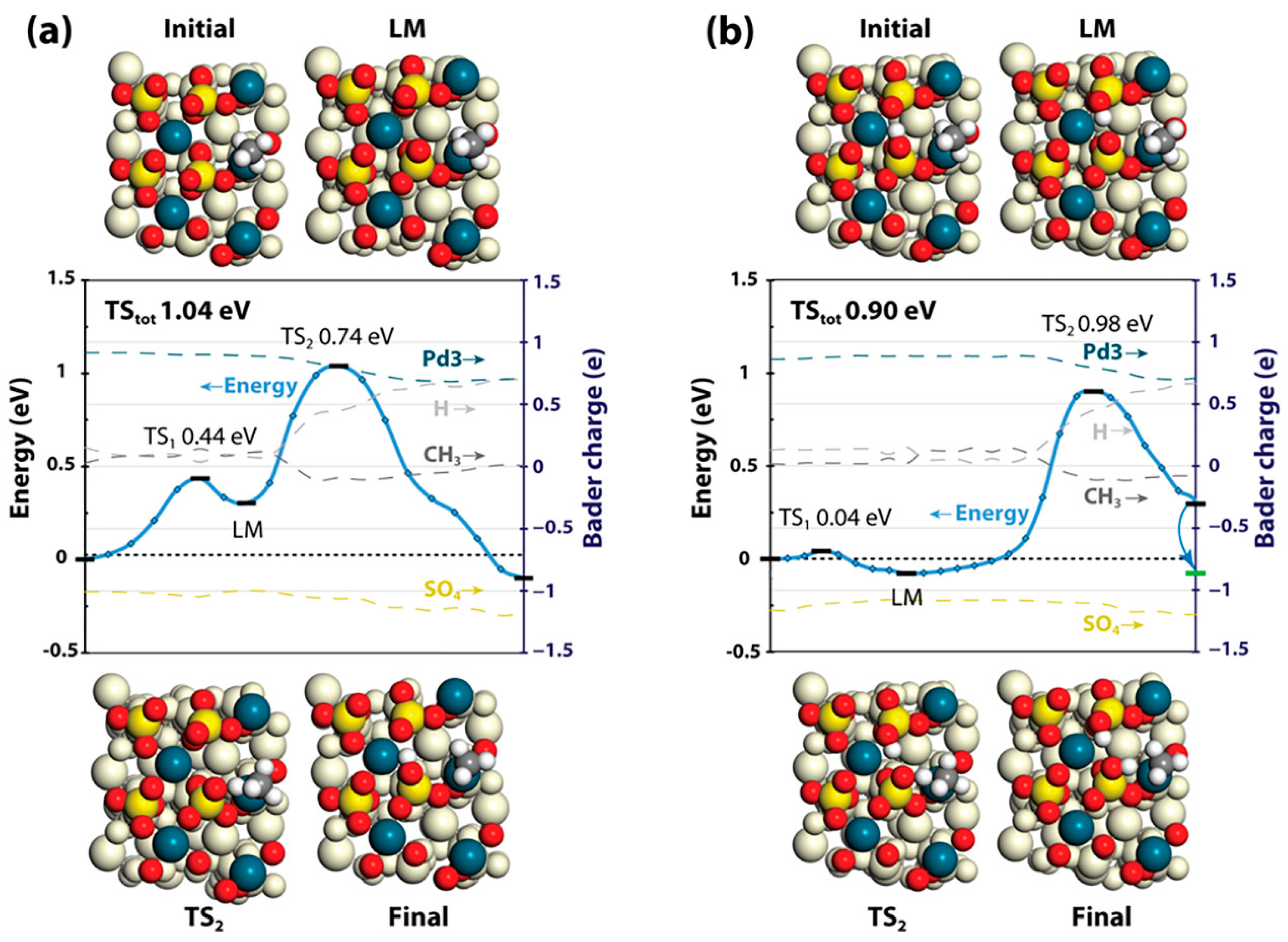
In actuality, most of the catalysts are unavoidably poisoned by a trace amount of SO
2 or accumulated sulfur under actual reaction conditions, which, hence, impairs the overall catalytic efficiency of the after-treatment system. In essence, the interaction between SO
2 and supported noble metal catalysts is a heterogeneous reaction of SO
2 with the active site or support, in which rich −OH groups, active oxygen species, and defects on the catalyst surface provide the necessary sites for SO
2 adsorption and transformation [
110]. Suvanto et al. studied the thin PdSO
4 films formed on the Pd–Pt/Al
2O
3 catalyst surface using the DFT calculations, and found that PdSO
4(110) and PdSO
4(111) which corresponded to PdO(100) and PdO(101) were the most possible poisoned index surface [
111]. The first C–H dissociation of CH
4 on PdSO
4(111), which contained the coordinatively unsaturated palladium, was a favorable option, with an energy barrier of 0.74–0.87 eV similar to that of the metallic Pd
0 reported before (
Figure 16). Moderate activation energies offered reasonable understanding of appropriate methane conversions over PdSO
4 under the dry conditions. Unfortunately, the presence of −OH groups was found to enhance the C–H dissociation barrier to terminal oxygen by 0.2 eV compared with the hydroxyl-free surface. Furthermore, water adsorption on PdSO
4(111) with an energy of −1.45 eV (−140 kJ/mol) was significantly stronger than that on PdO(101) with an energy of −0.82 eV (−79 kJ/mol), which occupied dominant competitive adsorption to bring about site blocking. The −OH groups weakened the adsorption on PdSO
4(111), while strong adsorption energy of the −OH groups was thermodynamically favorable for forming adsorbed molecular water on the catalyst surface. The presence of surface −OH groups was responsible for the increased C–H dissociation barrier, strong H
2O adsorption, and associated site blockage, which revealed the observation that the PdSO
4/Al
2O
3 sample poisoned by SO
2 under the humid conditions showed low methane conversions. Noble-metal-based materials with SO
2 intoxication gave rise to the surface sulfur deposition with the decreased methane oxidation activity. Moreover, the SO
2-poisoned catalysts facilitated rearrangement of the crystal structure and modified electronic properties of the noble metals.
Previous investigation results indicated that SO
2 adsorption induced recrystallization of the Pt(111) facet to the Pt(100) facet via surface diffusion driven by the decreased surface free energy of Pt/Al
2O
3 [
112,
113]. In particular, the SO
2-poisoning effect was intensively investigated on the reactions (e.g., ring opening of methyl cyclopentane, hydrogenolysis, and isomerization) which were sensitive to surface structures of the catalysts as well as sulfur poisoning [
112]. The sulfur species possessing an electronegative character could decrease electronic density of the Pt clusters, thus suppressing the adsorption ability of Pt toward the other adsorbates [
114]. Recently, Peng et al. [
97] developed an efficient strategy to synthesize the Pd–Ce nanowire-like catalysts with the good SO
2 tolerance in CH
4 combustion using the enhanced metal oxide interaction and core–shell confinement methods (
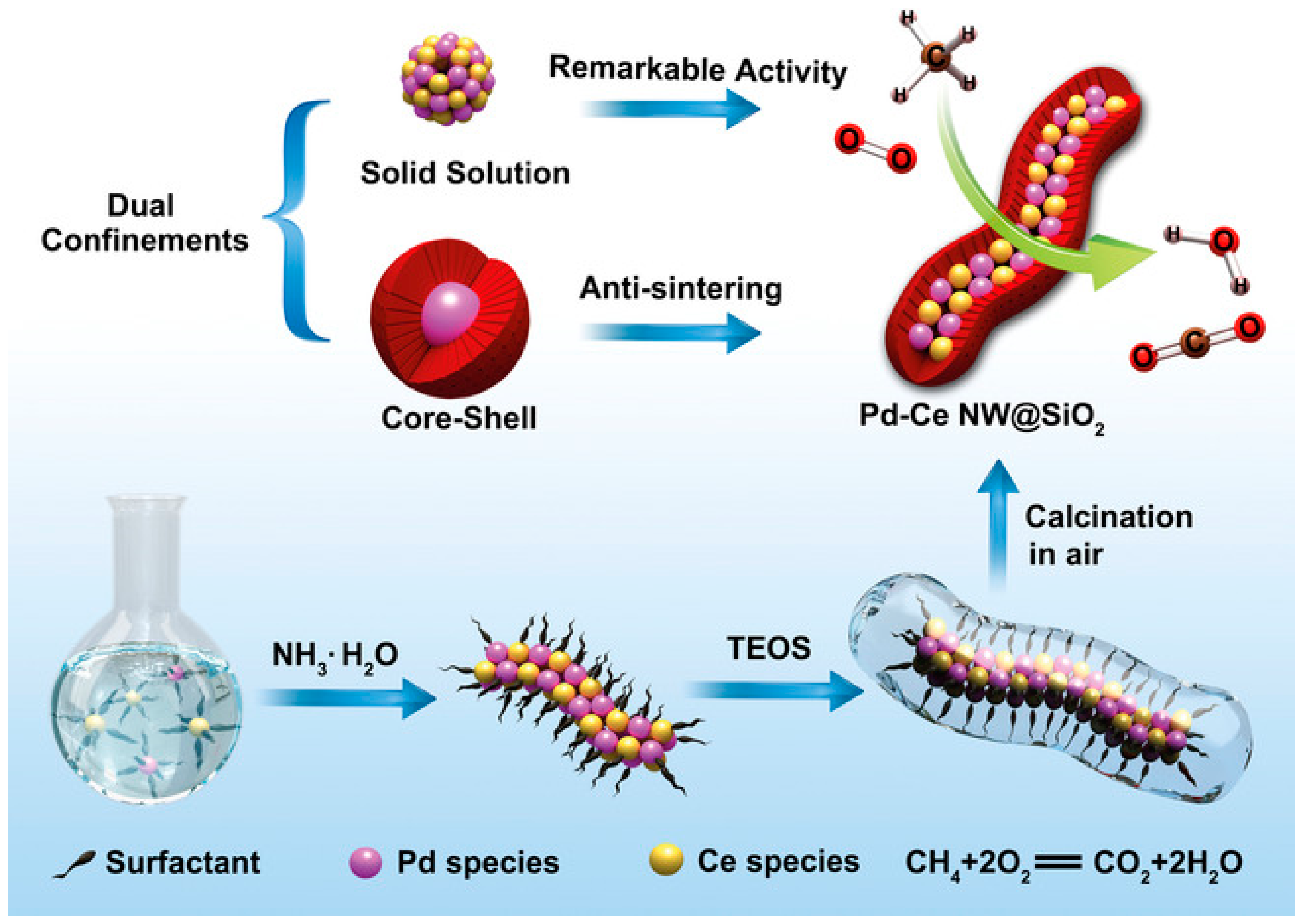
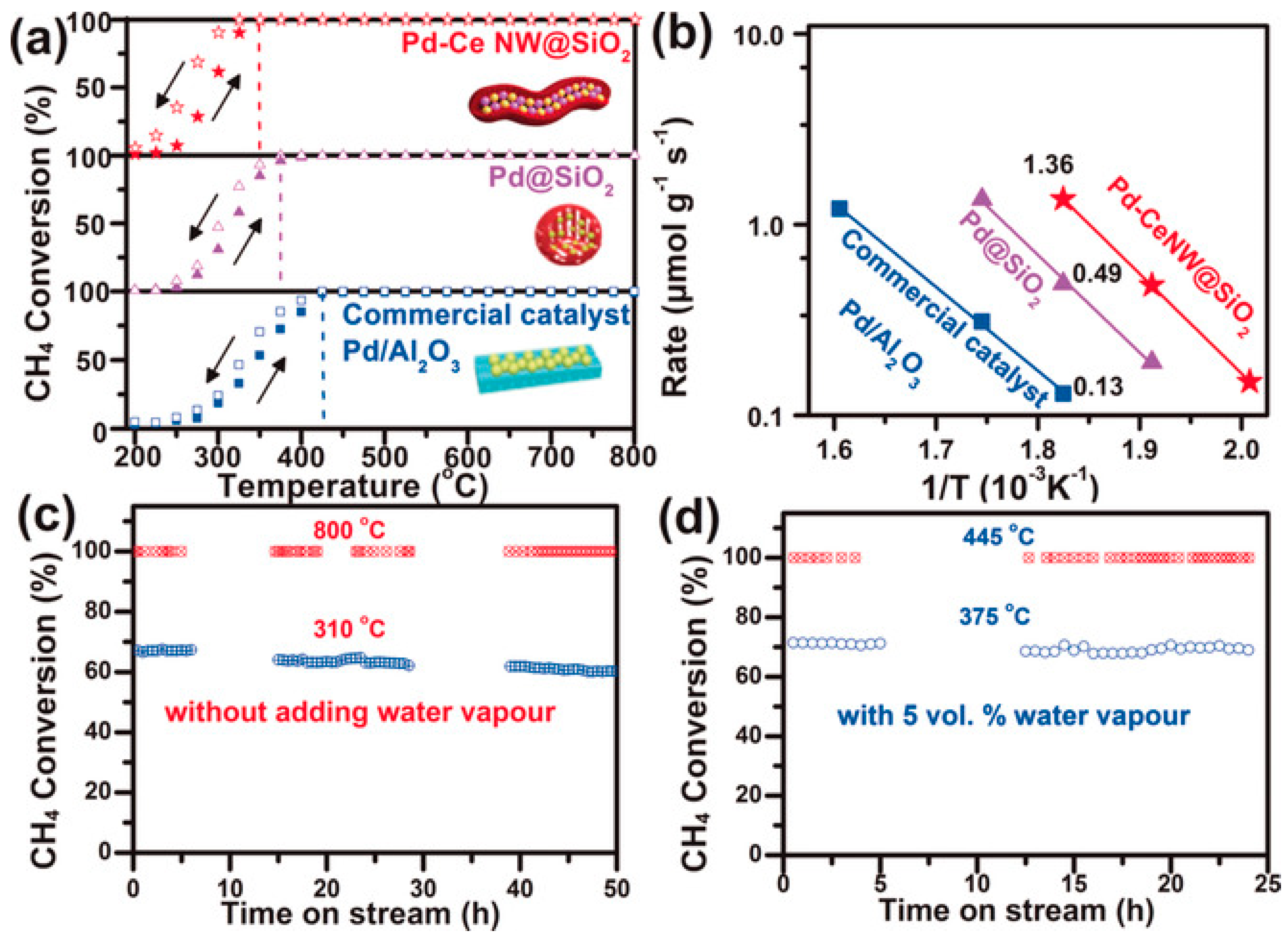
Figure 17 and
Figure 18). The as-prepared Pd–CeNW@SiO
2 sample exhibited complete CH
4 combustion within 10 h of on-stream reaction in the simultaneous presence of sulfur dioxide and moisture at 450 °C. In situ DRIFTS spectra of SO
2 adsorption displayed featured bands at 1255 and 1110 cm
−1 assignable to PdSO
4, which was considered to be the culprit for deactivation of the Pd-based catalysts. Fortunately, the porous shell structure of Pd–CeNW@SiO
2 exerted a shielding effect of SO
2 poisoning and a protection role for CeO
2 on Pd, which accounted for the excellent SO
2 tolerance.




















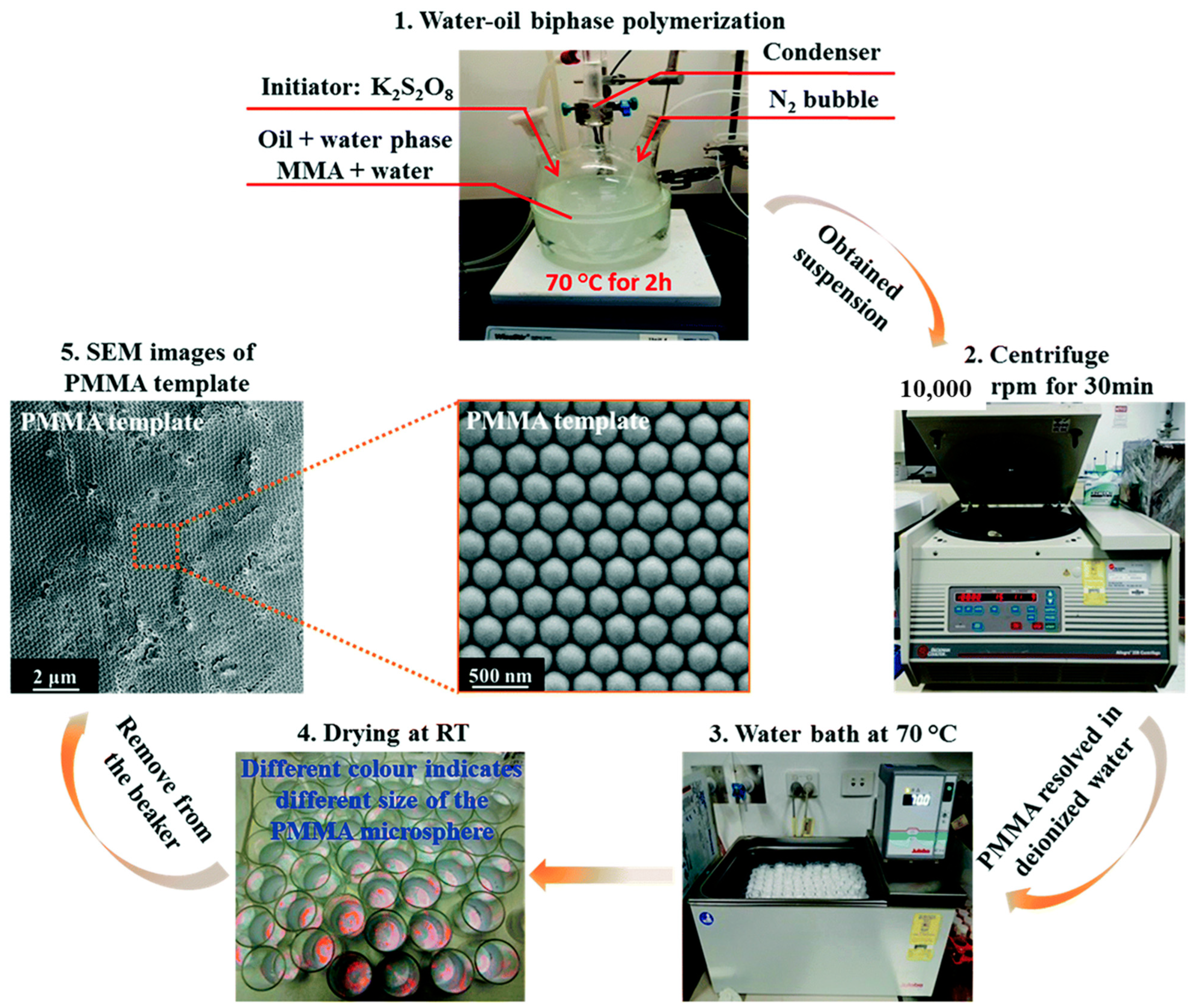{kind=link}
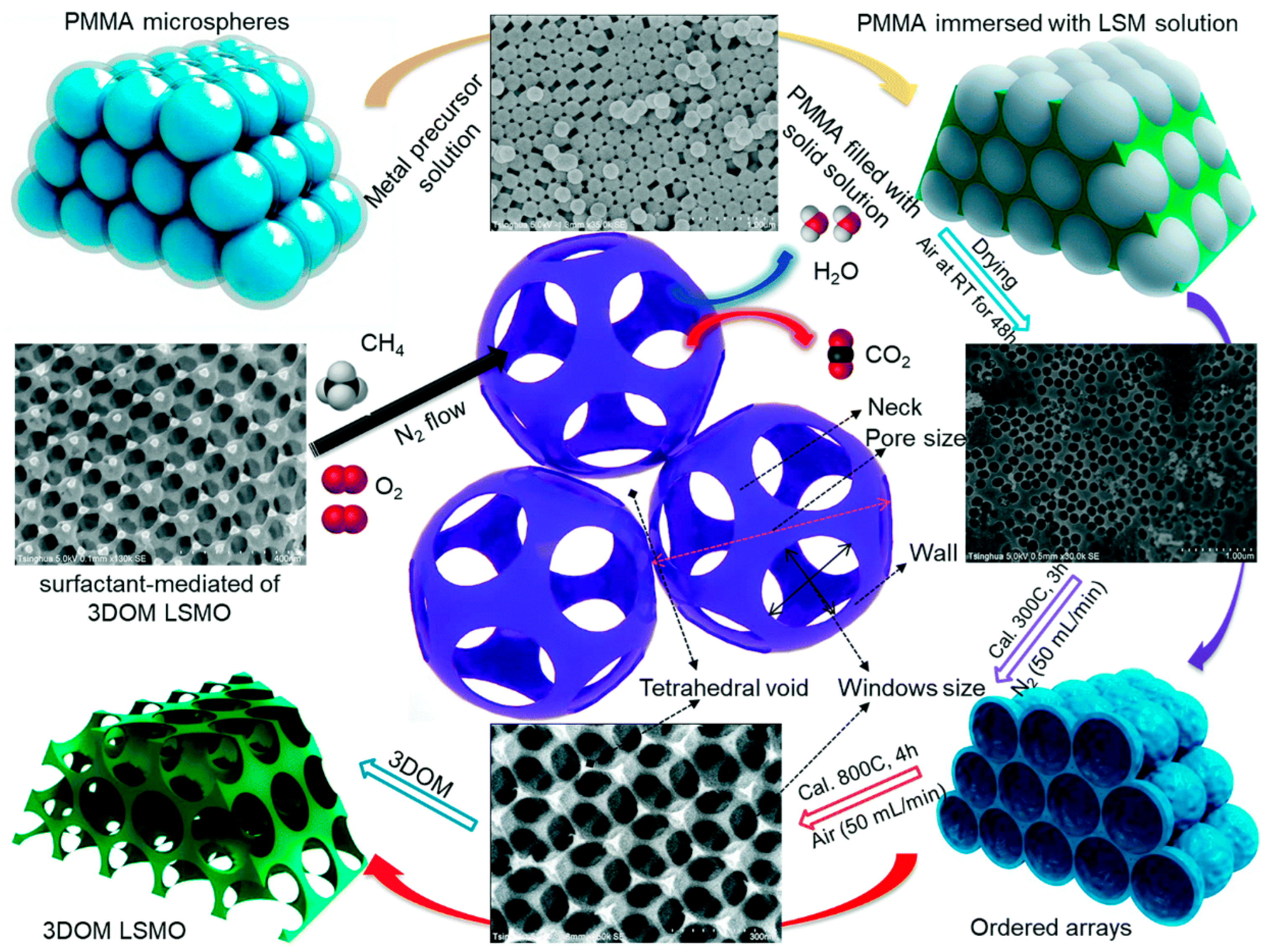{kind=link}
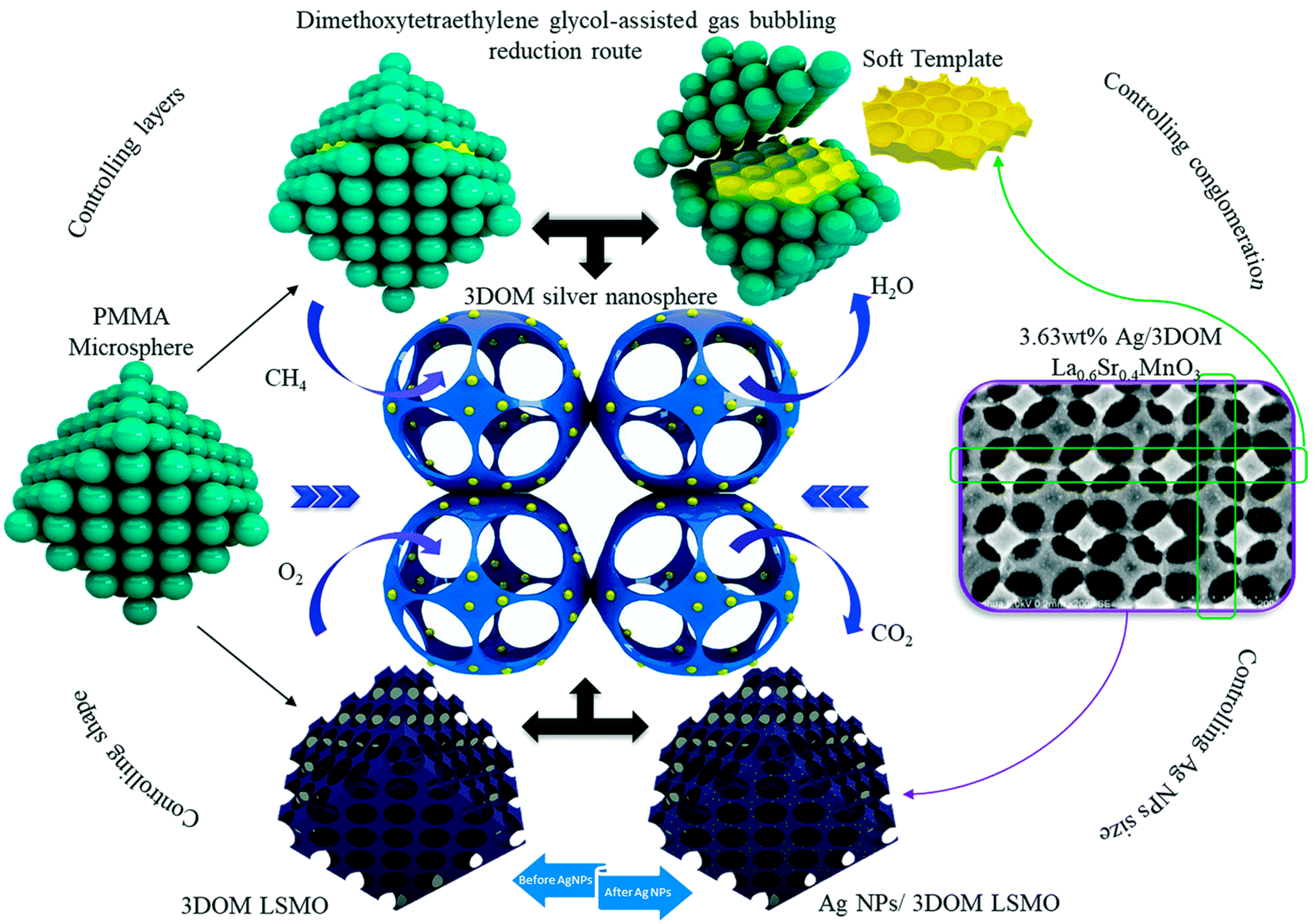{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}