1. Introduction
Intramolecular electronic communication between electrochemically active groups connected by a bridging moiety can be modified through small changes in the spatial disposition of the redox active moieties and/or by the nature of the central core. In these compounds, the bridging moiety has a central role in the spatial arrangement of the redox moieties, in addition to being a vital component in electron/hole transfer (e.g., the Creutz-Taube ion [Ru
2(NH
3)
5(pz)]
5+, pz = pyrazine) [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13]. Moreover, changes in the geometry and electron structure of the bridge generally affect the ability for electronic communication between the two redox moieties attached [
1,
2,
3,
4,
5,
6,
10,
11,
14]. In these compounds, the bridging ligand often contains either low energy π*-orbitals or high energy π-orbitals, to allow electrons or electron holes to freely move between the redox centers. To obtain electronic communication between redox centers, chirality (i.e., the use of a chiral bridging unit) can be successfully employed, to control the absolute geometry of the molecule and thus to direct and affect the communication between two (or more) redox centers [
14,
15].
During the last few decades, environmental issues and the depletion of petroleum resources turned chemists’ attention toward the exploitation of renewable feedstocks. In this context, following the rise of green chemistry, different biomasses were studied, and, in a short space of time, various waste products became potential alternative materials.
Focusing on chiral bio-based materials, among compounds belonging to the
chiral pool, a great deal of attention was addressed to isohexides, namely isomannide and isosorbide, used mainly in the polymer, cosmetic, and pharmaceutical industries (
Figure 1) [
16,
17,
18,
19,
20,
21].
Isomannide
1 and isosorbide
2, or (3R,3R,6R,6aR)-hexahydrofuro [3,2-b]furan-3,6-diol and (3R,3R,6S,6aR)-hexahydrofuro[3,2-b]furan-3,6-diol, respectively, obtained from the dehydration of D-mannitol and D-sorbitol, are important by-products of the starch industry and the processing of corn oil [
22]. These commercially available starting materials provide easy and inexpensive access to optically pure functionalized compounds, [
23,
24,
25] mainly exploiting the two hydroxyl groups at C3 and C6, both
endo in isomannide
1, whereas OH at C6 is
exo and OH at C3 is
endo in isosorbide
2 (
Figure 1). These functional groups can be easily derivatized, to obtaining a plethora of derivatives whose properties depend both from the characteristics of the introduced functional groups and from the different stereochemical arrangement of the hydroxyl groups [
23,
26,
27,
28,
29].
In previous studies [
30,
31,
32,
33,
34,
35], some of us successfully employed these compounds as starting materials for the synthesis of new chiral auxiliaries [
30,
31,
32,
33] and light-emitting materials [
34]. These studies highlighted the importance of the chiral backbone, which leads to an interaction between the functional groups linked to the central scaffold, with a great influence on the final properties, both in isomannide
1 and isosorbide
2. Moreover, some peculiar derivatives showed interesting applications in electrochemistry-related studies [
31].
Starting with these results, and prompted to expand on the applications of these interesting chiral bio-based compounds, we focused our attention on designing new materials for electronic communication.
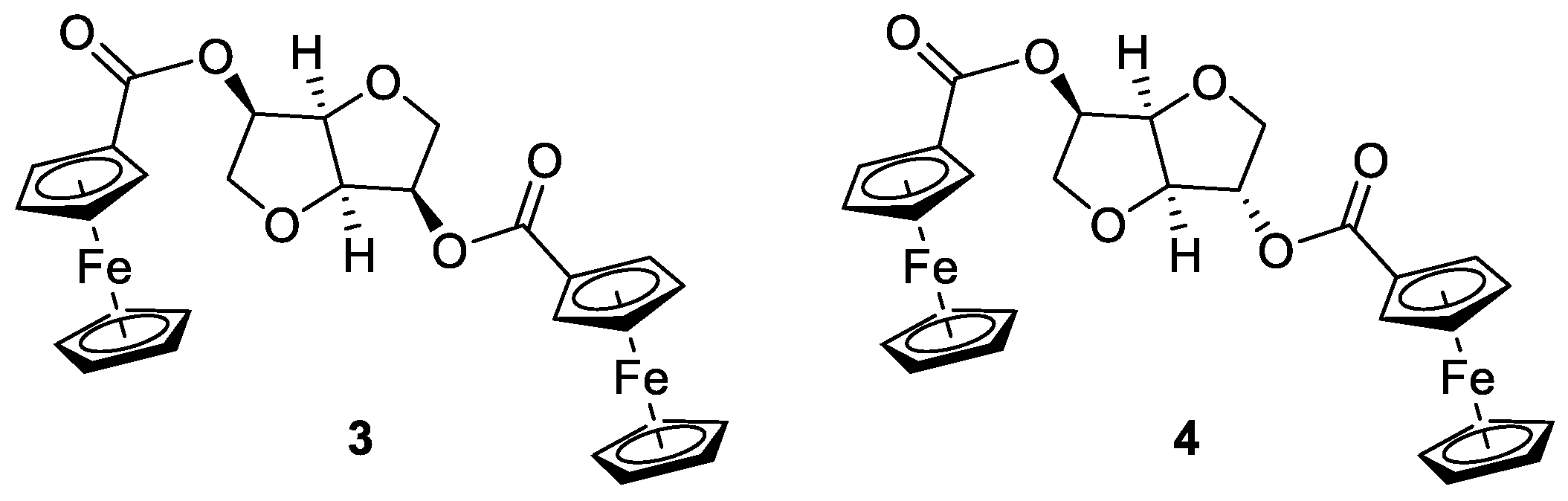
In this study, starting from isomannide
1 and isosorbide
2, the corresponding ferrocenyl diesters
3 and
4 were easily synthesized (
Figure 2). These new derivatives were selected to have easy access to isohexide-containing compounds characterized by the presence of two ferrocenyl moieties spatially close within the same chiral molecule (i.e., at least in principle). We focused our attention on derivatives
3 and
4 due to their ease of preparation, by simple double esterification of the free hydroxy groups, and to gain a deeper insight into the different dispositions of the two ferrocene moieties around the central chiral scaffold, an interesting feature to have a deeper insight in the influence of stereochemistry on electron communication. Compounds
3 and
4 were synthesized following the known protocols developed for the preparation of other isohexide diesters [
30,
31,
36]. These new derivatives, obtained as crystalline solids, were fully characterized through X-ray diffraction analysis (XRD) to gain more insight into their properties in the solid state. Then preliminary electrochemical and spectroelectrochemical studies were performed to assess the presence of electrochemical communication between the two ferrocenyl electroactive moieties.
The experimental results were supported by a time-dependent DFT (TD-DFT) study.
3. Results and Discussion
3.1. Synthesis of Ferrocenyl Derivatives
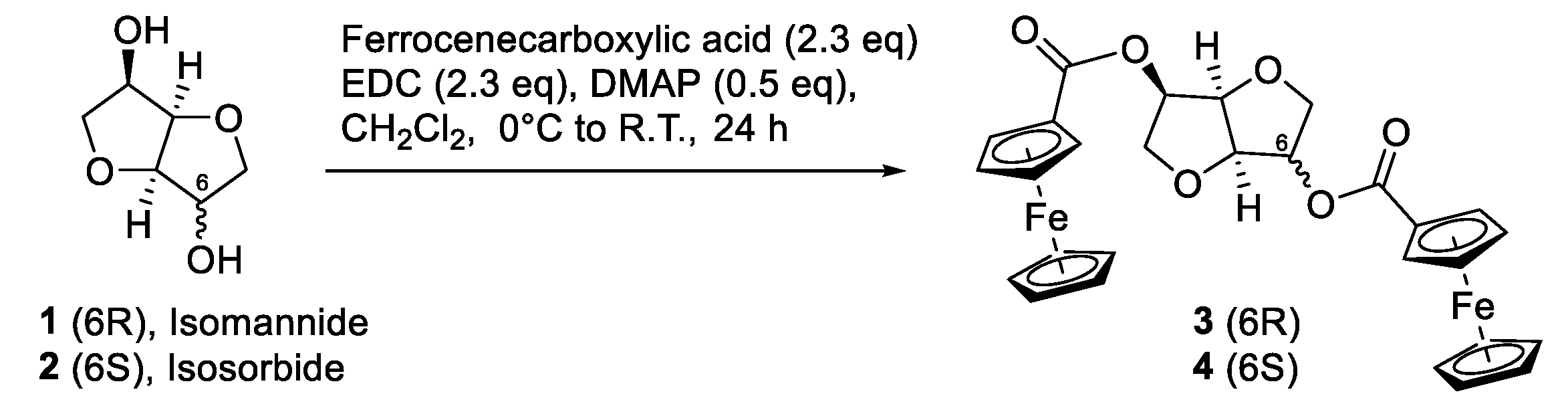
The ferrocenyl (Fc) derivatives of the isohexides were synthesized as reported in
Scheme 1. The two bis-ferrocenyl derivatives
3 and
4 were obtained in high yield by reacting isomannide
1 and isosorbide
2, respectively, with ferrocenecarboxylic acid in the presence of N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and N,N-4-dimethylaminopyridine (DMAP).
3.2. Crystal Structure of Ferrocenyl Derivatives 3 and 4
The ferrocenyl (Fc) derivatives
3 and
4 were obtained as orange crystals after recrystallization from hexane/ethyl acetate. The analysis of their structure through X-ray diffraction (XRD) showed that both
3 and
4 crystallized in chiral space groups, with
3 crystallizing in
P2
12
12 space group while the isosorbide derivative
4 crystallized in the space group
P2
12
12
1 (
Figure 3). More details regarding the pertinent metric parameters of the crystal structures are reported in the
Supporting Information (Section 1.3).
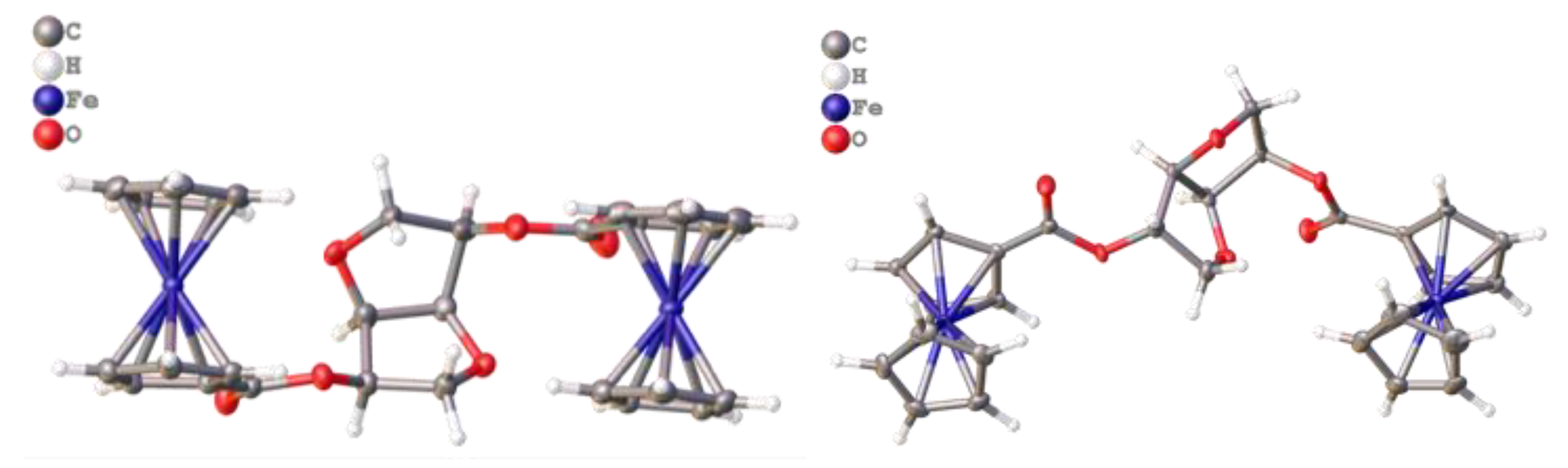
The molecular structure of
3 obtained from XRD, showed that the two Fc moieties are oriented parallel, one to the other, around the central chiral scaffold with respect to their Cp-Fe-Cp axis (
Figure 3,
left). Moving to the crystal structure, the results showed that each molecule of
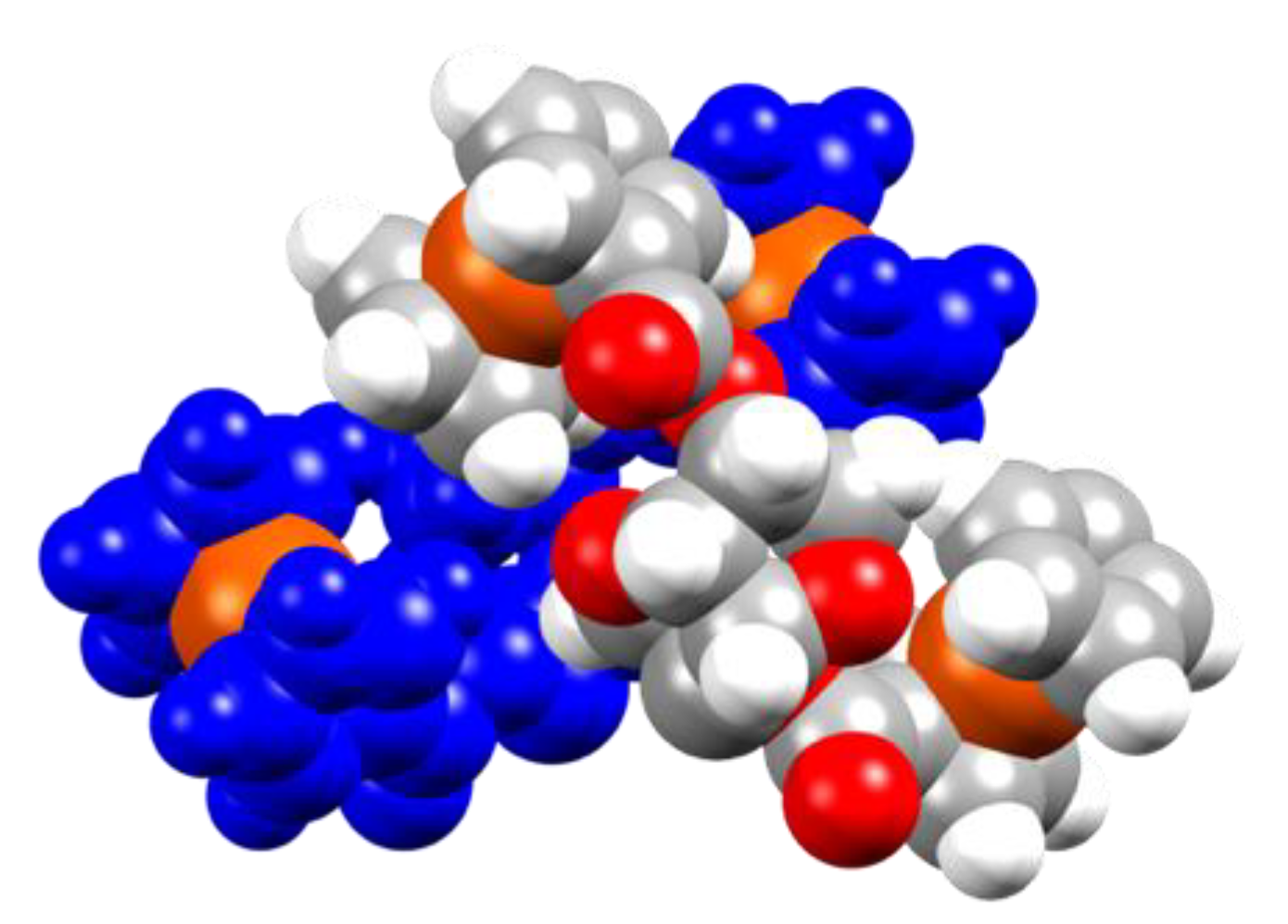
3 packs with one neighboring molecule, with one of the Fc moieties of the second molecule of
3, entering the chiral cavity of the other molecule orthogonally to its symmetry (i.e., the Cp-Fe-Cp axis of the two molecules are one orthogonal to the other and one of the Fc moieties of the second molecule of
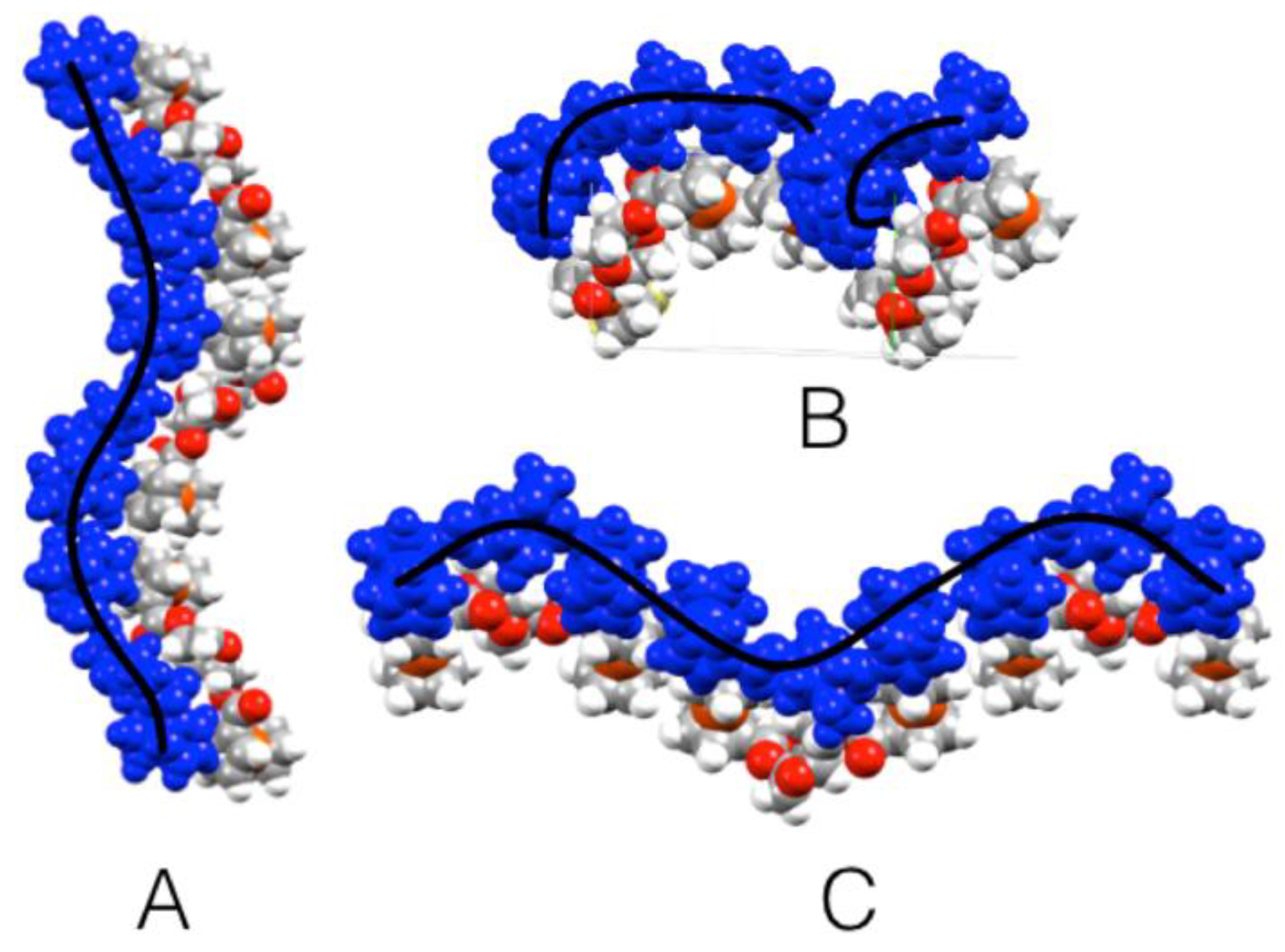
3 is located in between the chiral cavity and the Fc units of the first one). The second Fc moiety is located outside the chiral pocket generating a V-type dimer, as shown in
Figure 4. The next layer of molecules forms a helical structure along the
b-axis (
Figure 5).
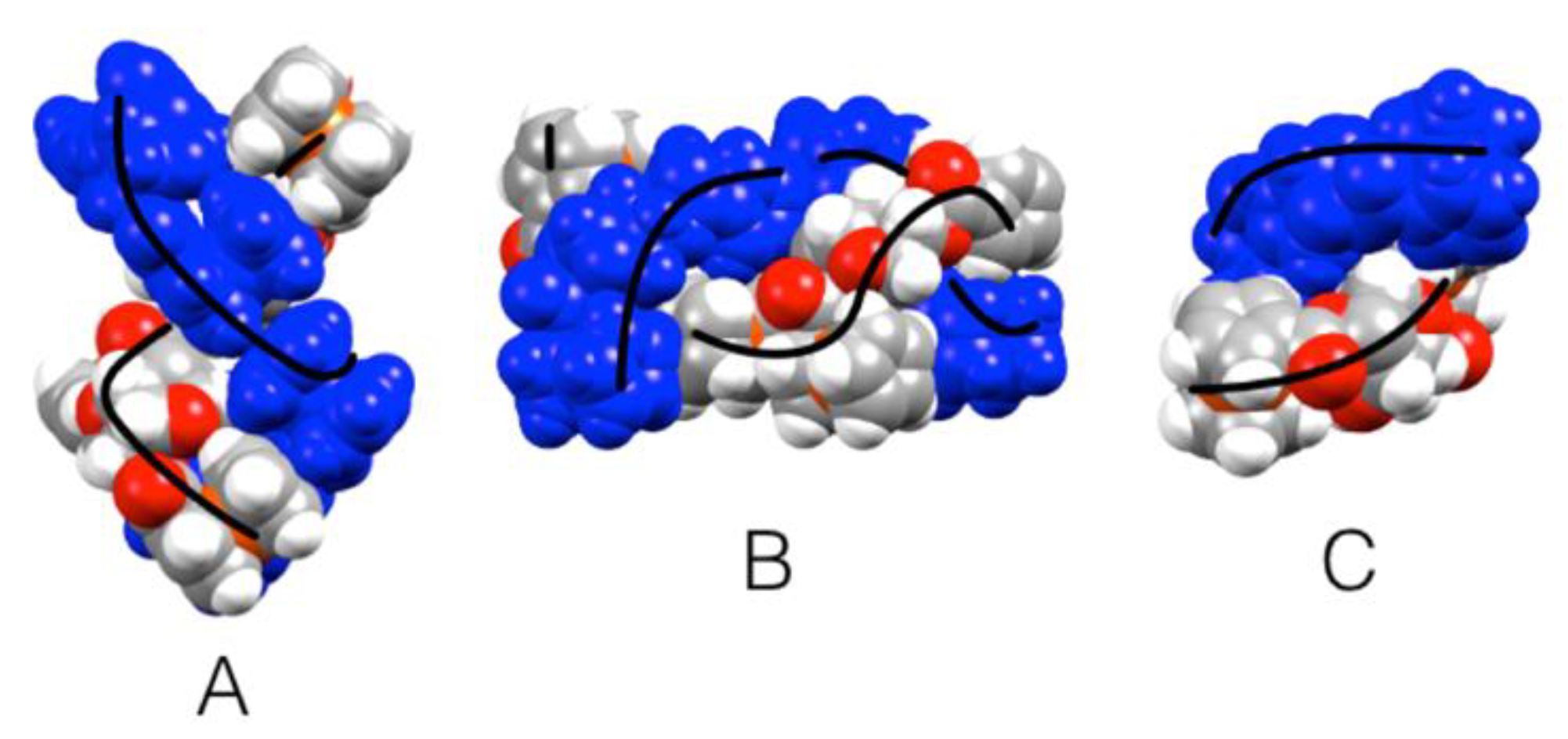
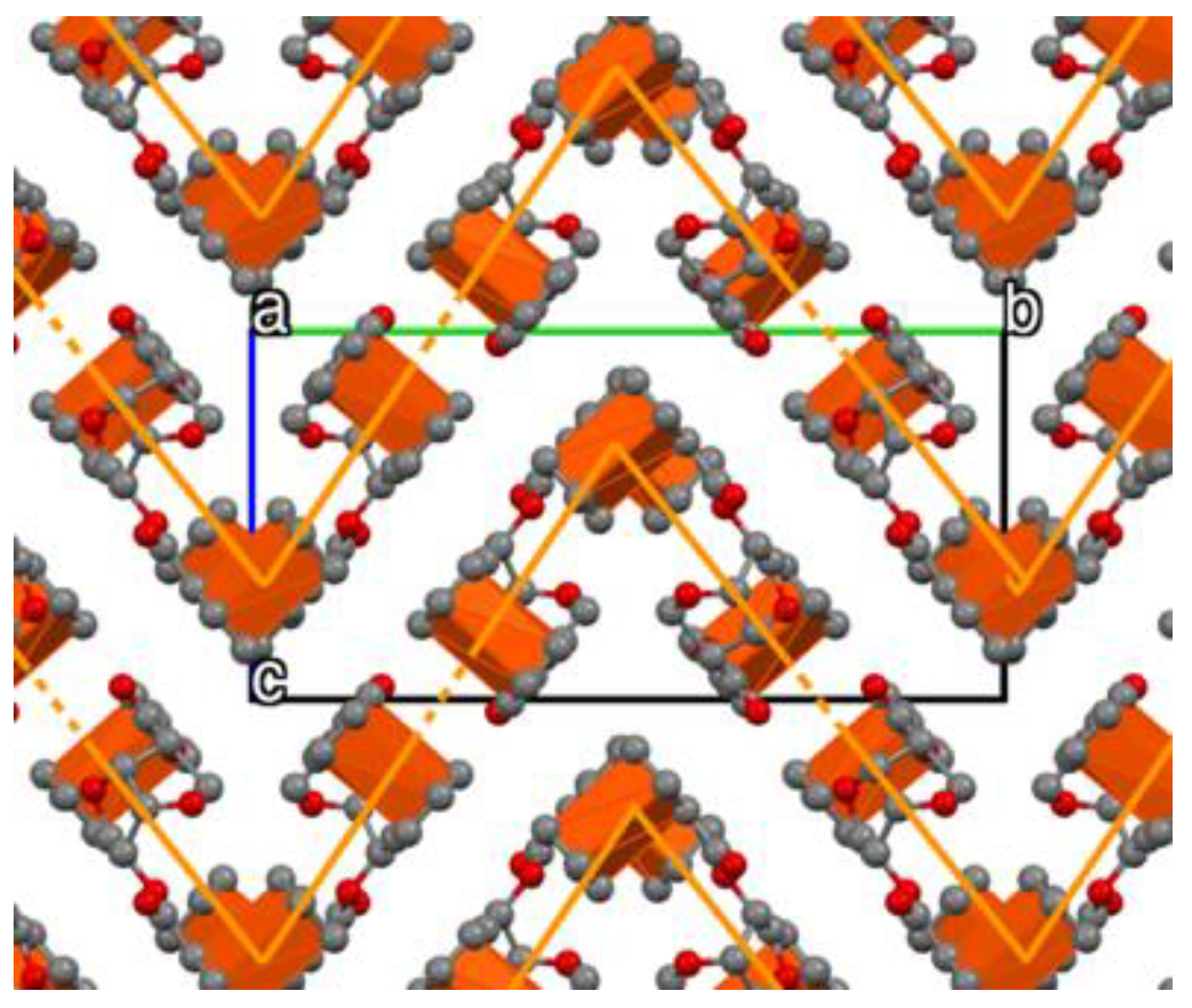
The crystal packing of
3 forms a zig-zag pattern generated from the V-shaped dimers that are repeated along the
c-axis, with the following layer along the
b-axis being rotated at 180° (
Figure 6). The crystal of
3 forms a tight molecular staggering following the
a-axis. Looking down the
c-axis the staggering of the layers formed by the Fc moieties could be easily seen (
Figure 6). This crystallographic feature of the Fc moieties is controlled by the geometric constraints imparted by the chiral isomannide bridge due to the orientation of both Fc moieties on the same side of the molecule (i.e., the two Fc moieties are located on the same side of the chiral cavity generated by the fused bicyclic structure). These geometric constraints generate a sort of pocket that allows two molecules to form a supramolecular dimer, and that lead to the extended helical supramolecular packing along the
b-axis shown in
Figure 5. The Fc moieties showed an orthogonal C-H to Cp face approximation (e.g., CH approximation to the
π system of adjacent molecules), similar to C-H to π-stacking observed in classic aromatic systems [
43].
In contrast to
3, the molecular structure of
4 showed that one of its Fc moieties is oriented above both the planes of the two fused tetrahydrofuran cycles of the molecule, while the other Fc is oriented almost in the plane of the neighboring five-term cycle. More clearly, the two Fc units are located on opposite sides of the chiral cavity generated by the fused bicyclic structure of isosorbide. This geometric change from the isomannide bridge in
3 to the isosorbide bridge in
4 leads to the intertwining of adjacent molecules since they lack the pocket found in the packing of
3 (
Figure 4 and
Figure 5). The configuration of the isosorbide bridge in compound
4 resulted in an open helical structure along the
b-axis, as depicted in
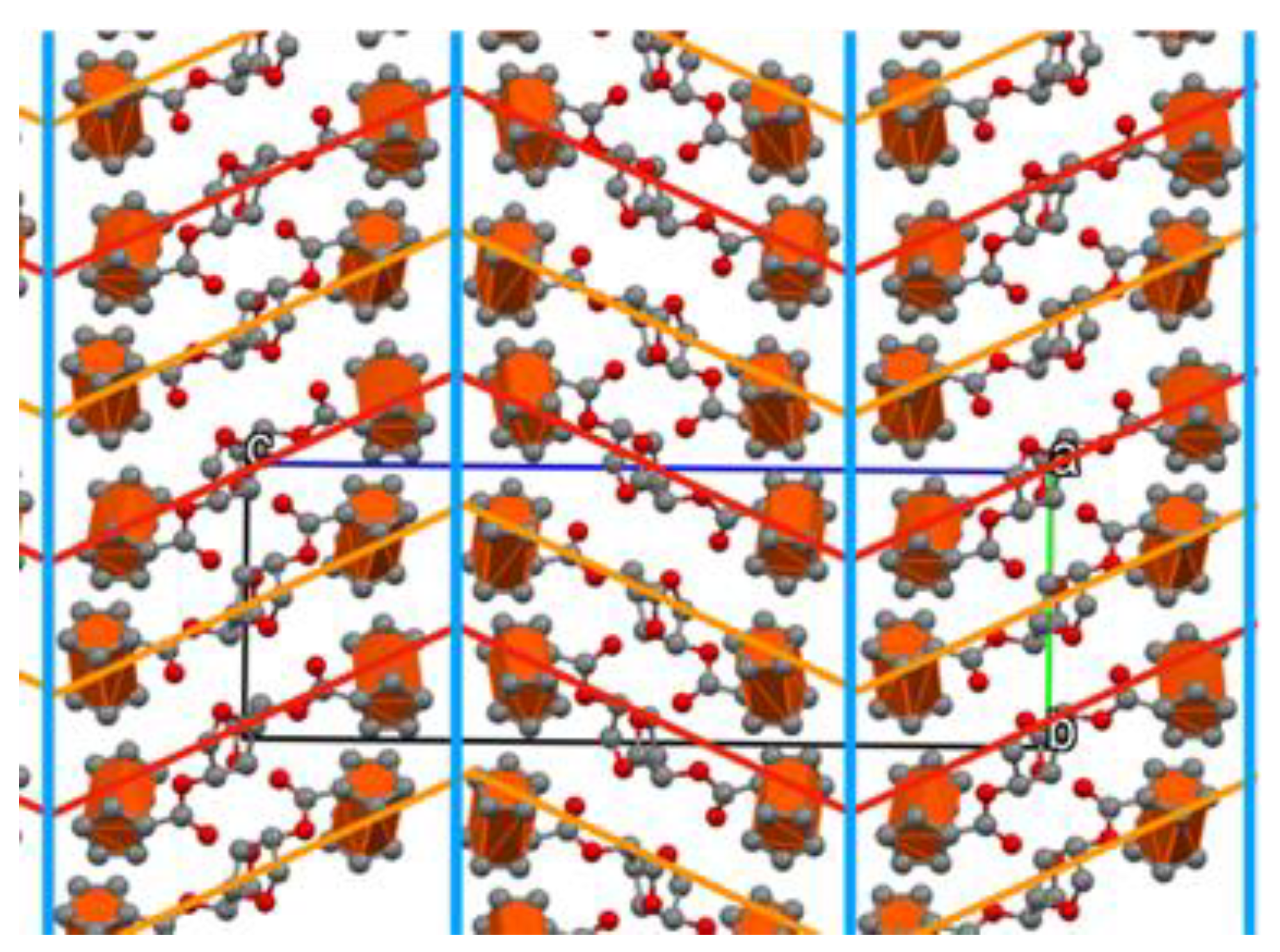
Figure 7. Looking down the
a-axis of
4, the molecules form a much looser zig-zag pattern where the Fc moieties are oriented parallel to their neighboring partner along the
c-axis, and with its Fc moieties packing orthogonally along the
b-axis (
Figure 8). The Fc moieties are also aligned to form different “sheets”, highlighted in light blue in
Figure 8, along the
b-axis, where each Fc moiety is oriented orthogonal to the neighboring Fc moiety in the next molecule, similar to the C-H to Cp face interaction observed in
3. Looking down the
b-axis, the sheets of Fc moieties are also oriented along the
a-axis (highlighted in red in
Figure 8). The particular and diverse orientation of the two Fc moieties in compound
4, described above, results in a more open packing where the Fc moieties could arrange in a series of planes that stack one over the other (
Figure 7).
3.3. Electrochemical and Spectroelectrochemical Studies of Compounds 3 and 4
Once the molecular structure and the crystal structure of compounds 3 and 4 were fully characterized, electrochemistry and spectroelectrochemistry were employed to determine the role of the different orientations of the two Fc moieties in 3 and 4 on their electronic communication.
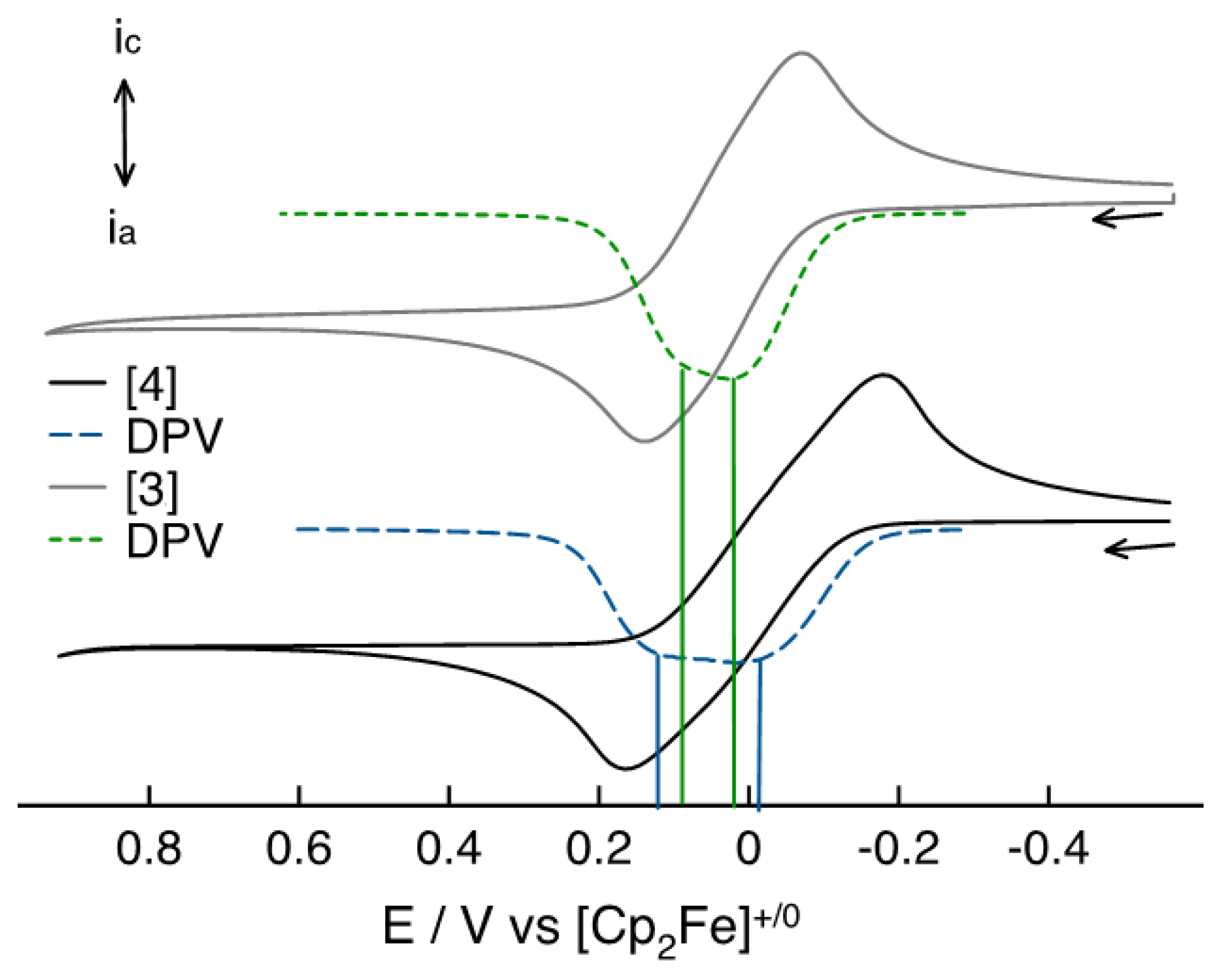
The analysis, made through cyclic voltammetry (CV) and differential pulsed voltammetry (DPV), showed the presence of two redox processes for both
3 and
4, with a Δ
E = 77 mV for
3, consistent with a comproportionation constant
Kc = 10
1.6, and a Δ
E = 154 mV for compound
4, highlighting a
Kc =10
2.6, an order of magnitude larger (
Figure 9) [
23].
One of the reasons to consider justifying this difference could be represented by the non-equivalency, in terms of molecular symmetry, between the two Fc units in compound 4, due to the different stereochemistry of the original OH groups in isosorbide 2, whereas for compound 3 the Fc moieties are symmetry equivalent.
Whilst electrochemically determined half-wave potential splitting is inconclusive with respect to the strength of the electronic coupling in the mixed-valent state, it does indicate that the orientation of the Fc moieties does influence the final electric properties; surprisingly 4 shows a larger Kc despite the larger spatial separation of the two Fc moieties when attached to isosorbide as the central chiral scaffold.
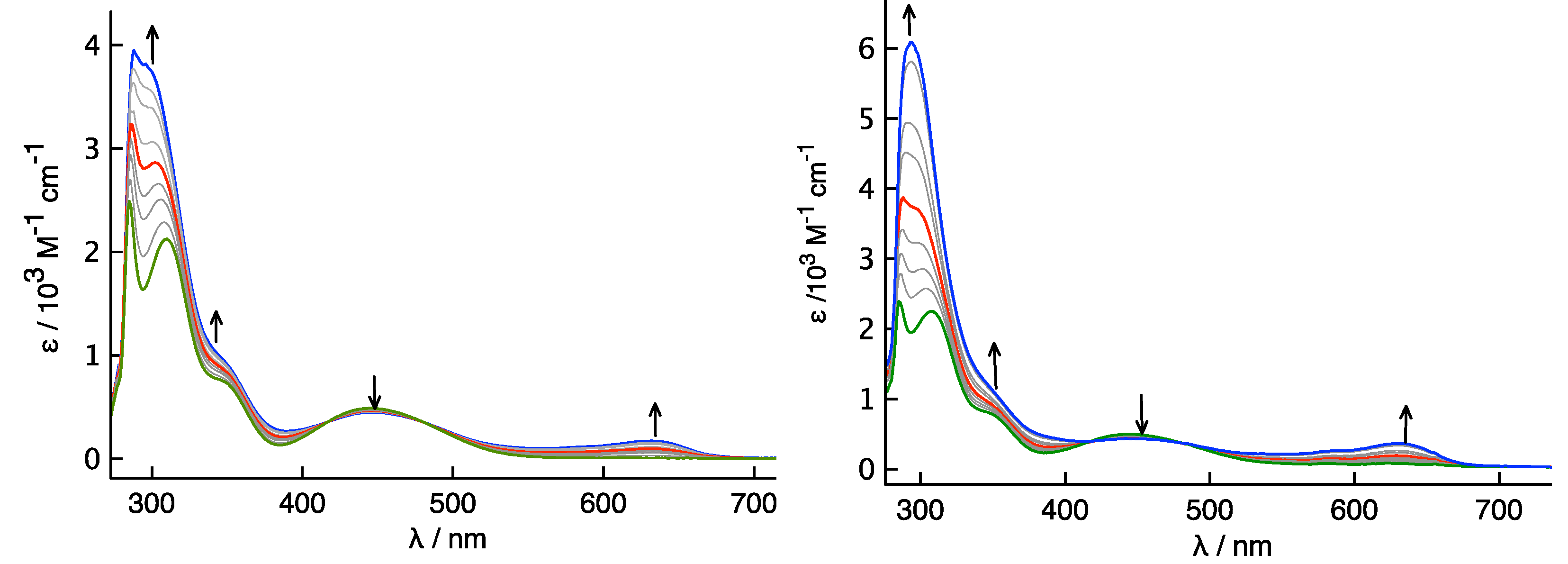
To gain a deeper insight into the origin of the larger Kc observed for 4, with respect to the one observed for compound 3, a UV-Vis NIR spectroelectrochemistry (SEC) study and TD-DFT calculations were performed.
The UV-Vis NIR spectroelectrochemistry (SEC) of
3 (
Figure 10 left) showed a band at λ
max = 635 nm that appeared upon oxidation. The second oxidation did not result in any quenching of this band, suggesting that the electronic transition associated with this band was related to the presence of the ferrocenium ion and not to the presence of an intervalence charge transfer (IVCT). This band, however, is characterized by lower energy than might be expected considering only an Fc
+ d-d excitation, due to an increased ligand to metal charge transfer (LMCT) deriving from the presence of the ester group on the Fc moiety, as showed by the time-dependent DFT (TD-DFT) analysis. Moving from the first to the second oxidation, some minor changes in the spectrum of
3+ and
32+ were observed, but the major one was represented by the change in the intensity of the band at 303 nm. Accordingly, the results obtained in the SEC analysis indicate that the process is fully reversible.
Despite their spatial proximity, the results showed a lack of communication between the two Fc redox moieties in isomannide derivative 3; this behavior could be related to the restricted communication between the Fc+ ion and the chiral bicyclic bridge.
While the molecular structure of
4 in the crystal state showed that both Fc moieties were oriented further apart, the CV suggested a higher degree of intermolecular electronic communication, as highlighted by the higher value of the
Kc = 10
2.6, an order of magnitude higher than the one obtained for derivative
3. UV-Vis SEC was employed again to determine if the change in orientation of the two Fc moieties does affect the degree of electronic communication. The two oxidation processes could be separated in the UV-vis SEC, as seen in
Figure 10 (
right side). Oxidation to
4+ showed a similar low energy band at l
max = 630 nm, as observed in the UV-vis NIR SEC spectrum of
3+, and this CT was also due to a LMCT from the ester to the electron-hole on the iron. Further oxidation to
42+ only caused an increase in the intensity of the band at λ
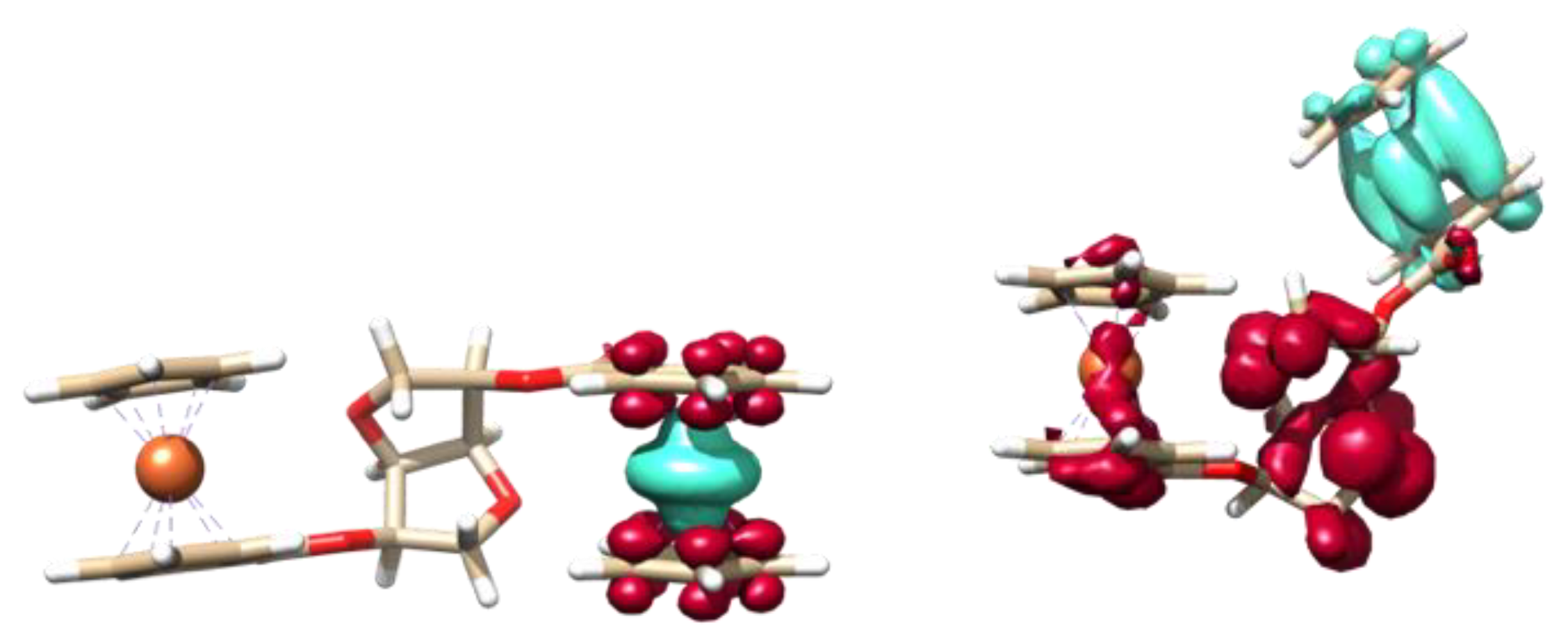
max = 630 nm, which again would indicate that this band is not an IVCT. TD-DFT was again used to help explain the transitions found in the UV-vis SEC spectrum. The lower energy bands are primarily due to LMCT from the Fe
II to the Cp-CO
2-R moiety, however, interestingly the Fc moiety in the plane of one of the rings of the bridge showed an increased charge transfer to the ether functionality in the isosorbide (
Figure 11 as well as some mixing from the Fe
II. This increased LMCT, calculated at 550 nm, from the isosorbide to the electron-hole showed that geometric control over the Fc moieties can be effectively exploited to increase electronic communication.
4. Conclusions
Bio-based chiral compounds, namely isomannide and isosorbide, were successfully employed to bridge two Fc moieties through simple derivatization of the starting materials. The use of different chiral bridges allowed us to obtain a different stereochemical disposition of the ferrocenyl units around the central scaffold. Furthermore, the different spatial dislocation of the Fc units was highlighted by the crystal structure of the two diesters 3 and 4. The analysis of the structures obtained by XRD showed that in compound 3, when isomannide was the central scaffold, the Fc moieties tend to be parallel, one to the other (considering their Cp-Fe-Cp axes), leading to the formation of a sort of chiral pocket and V-shaped intermolecular dimers. Conversely, when isosorbide was employed as a bridging scaffold, as for compound 4, an open structure was observed, with the helical supramolecular organization much looser. The electrochemical and spectroelectrochemical characterization of these compounds showed that in the mixed valent complex 4+ the electronic communication was slightly higher than in compound 3+ due to the ability of the Fc moiety to interact with the ether moiety in the isosorbide bridge. Moreover, these results were confirmed by TD-DFT studies. In conclusion, isomannide and isosorbide ferrocenyl derivatives 3 and 4 were shown to possess interesting structural and electronic properties, suggesting future possibilities for obtaining interesting new compounds by choosing not only the different moieties attached to the same central scaffold, but by exploiting the different stereochemical features of these two bio-based, easy to derivatize, cheap chiral scaffolds.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











