Local Effects of Ring Topology Observed in Polymer Conformation and Dynamics by Neutron Scattering—A Review


Abstract
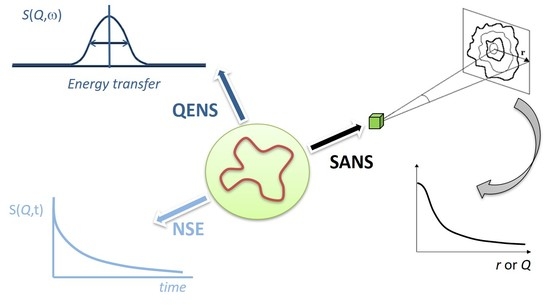
:
1. Introduction
2. Neutron Scattering
3. Cyclic Polymers
3.1. Conformation of Cyclic Polymers in Solution
3.2. Conformation of Cyclic Polymers in the Pure Melt State
4. Dynamics of Cyclic Polymers
4.1. Neutron Spin-Echo Studies
4.2. Quasielastic Neutron Scattering
5. Blends of Polymers with Different Topologies
5.1. Miscibility and Conformation
5.2. Dynamics
6. Summary: Experimentally Confirmed Theory and Open Questions
6.1. Conformation, Scaling, and Relative Sizes of Rings and Chains
6.2. Local and Large Scale Dynamics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Symbols and Abbreviations
| A2 | virial coefficient |
| b | scattering length |
| c | concentration |
| d | tube diamter |
| D | diffusion coefficient |
| GPC | gel permeation chromatography |
| HPLC | high pressure liquid chromatography |
| I(Q) | scattered intensity |
| kB | Boltzmann constant |
| KWW | Kohlrausch-Williams-Watts function or stretched exponential |
| LCCC | liquid chromatography at the chromatographic critical condition |
| M | molecular weight of the polymer |
| Mc | critical molecular weight at which entanglements effects are first observed |
| Me | entanglement molecular weight |
| Mo | molecular weight of polymer repeat unit |
| Mw | weight average molecular weight |
| Mz | z-average molecular weight |
| msd | mean-square displacement |
| NA | Avogadro’s number |
| Ni | degree of polymerisation of polymer i |
| nn | number average number of bonds |
| NMR | nuclear magnetic resonance |
| NSE | neutron spin echo |
| OM | Optical microscopy |
| PB | poly(butadiene) |
| PDMS | poly(dimethyl siloxane) |
| PEO | poly(ethylene oxide) |
| PS | poly(styrene) |
| P(Q) | form factor |
| Q | scattering vector or momentum transfer |
| QENS | quasielastic neutron scattering |
| Rg | radius of gyration |
| Rz | z-average radius of gyration |
| Rw | weight-average radius of gyration |
| <r2cm(t)> | centre of mass mean square displacement |
| SANS | small angle neutron scattering |
| SLD | neutron scattering length density |
| S(Q) | structure factor |
| S(Q,t) | intermediate scattering law |
| S(Q,ω) | dynamic structure factor |
| T | temperature |
| Ts | spinodal point |
| Z | number of entanglements |
| β | stretched exponent |
| χ | segment-segment interaction parameter |
| φI | volume fraction of polymer i |
| η | melt viscosity |
| ηs | solvent viscosity |
| ηo | zero-shear rate viscosity |
| λ | wavelength |
| ν | Flory exponent |
| νi | volume of repeat unit |
| νo | reference volume |
| θ | scattering angle |
| density | |
| σ | spring length of the Rouse model |
| τ | characteristic time |
| τe | Rouse relaxation time at the entanglement length Ne |
| τeff | effective time |
| τR | Rouse relaxation time |
| τrep | reptation time |
| ω | frequency |
| Ω | inverse correlation time |
| ξ | correlation length |
| (Δρ)2 | neutron contrast factor |
References
- Arrighi, V.; Cowie, J.M.G. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Mark, J.; Ngai, K.; Graessley, W.; Mandelkern, L.; Samulski, E.T.; Koenig, J.; Wignall, G.D. Physical Properties of Polymers, 3rd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Kricheldorf, H.R. Cyclic Polymers: Synthetic Strategies and Physical Properties. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 251–284. [Google Scholar] [CrossRef]
- Ungar, G.; Zeng, K.B. Learning polymer crystallization with the aid of linear, branched and cyclic model compounds. Chem. Rev. 2001, 101, 4157–4188. [Google Scholar] [CrossRef]
- Su, H.-H.; Chen, H.-L.; Diaz, A.; Teresa Casas, M.; Puiggali, J.; Hoskins, J.N.; Grayson, S.M.; Perez, R.A.; Mueller, A.J. New insights on the crystallization and melting of cyclic PCL chains on the basis of a modified Thomson-Gibbs equation. Polymer 2013, 54, 846–859. [Google Scholar] [CrossRef]
- Clarson, S.J.; Dodgson, K.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethylsiloxane): 19. Glass-transition Temperatures and Crystallization Behavior. Polymer 1985, 26, 930–934. [Google Scholar] [CrossRef]
- Huang, D.H.; Simon, S.L.; McKenna, G.B. Chain length dependence of the thermodynamic properties of linear and cyclic alkanes and polymers. J. Chem. Phys. 2005, 122, 084907. [Google Scholar] [CrossRef]
- Dimarzio, E.A.; Guttman, C.M. The Glass Temperature of Polymer Rings. Macromolecules 1987, 20, 1403–1407. [Google Scholar] [CrossRef]
- Liu, X.J.; Chen, D.L.; He, Z.D.; Zhang, H.; Hu, H.Z. Molecular-weight Dependence of the Glass-transition of Cyclic Polystyrene. Polym. Commun. 1991, 32, 123–125. [Google Scholar]
- Gan, Y.D.; Dong, D.H.; Hogenesch, T.E. Effects of lithium bromide on the glass-transition temperatures of linear and macrocyclic poly(2-vinylpyridine) and polystyrene. Macromolecules 1995, 28, 383–385. [Google Scholar] [CrossRef]
- Santangelo, P.G.; Roland, C.M.; Chang, T.; Cho, D.; Roovers, J. Dynamics near the glass temperature of low molecular weight cyclic polystyrene. Macromolecules 2001, 34, 9002–9005. [Google Scholar] [CrossRef]
- Dodgson, K.; Bannister, D.J.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethyl siloxanes): 4. Bulk Viscosities. Polymer 1980, 21, 663–667. [Google Scholar] [CrossRef]
- Orrah, D.J.; Semlyen, J.A. Rossmurphy, S.B. Studies of Cyclic and Linear Poly(Dimethylsiloxanes): 27. Bulk Viscosities above the Critical Molar Mass for Entanglement. Polymer 1988, 29, 1452–1454. [Google Scholar] [CrossRef]
- Orrah, D.J.; Semlyen, J.A.; Rossmurphy, S.B. Studies of Cyclic and Linear Poly(dimethyl siloxanes): 28. Viscosities and Densities of Ring and Chain Poly(dimethyl siloxane) Blends. Polymer 1988, 29, 1455–1458. [Google Scholar] [CrossRef]
- Mills, P.J.; Mayer, J.W.; Kramer, E.J.; Hadziioannou, G.; Lutz, P.; Strazielle, C.; Rempp, P.; Kovacs, A.J. Diffusion of Polymer Rings in Linear Polymer Matrices. Macromolecules 1987, 20, 513–518. [Google Scholar] [CrossRef]
- Tead, S.F.; Kramer, E.J.; Hadziioannou, G.; Antonietti, M.; Sillescu, H.; Lutz, P.; Strazielle, C. Polymer Topology and Diffusion—A Comparison of Diffusion In Linear and Cyclic Macromolecules. Macromolecules 1992, 25, 3942–3947. [Google Scholar] [CrossRef]
- Cosgrove, T.; Griffiths, P.C.; Hollingshurst, J.; Richards, R.D.C.; Semlyen, J.A. Self-Diffusion and Spin Spin Relaxation in Cyclic and Linear Poly(dimethyl siloxane) Melts. Macromolecules 1992, 25, 6761–6764. [Google Scholar] [CrossRef]
- Cosgrove, T.; Turner, M.J.; Griffiths, P.C.; Hollingshurst, J.; Shenton, M.J.; Semlyen, J.A. Self-diffusion and Spin-spin Relaxation in Blends of Linear and Cyclic Poly(dimethyl siloxane) melts. Polymer 1996, 37, 1535–1540. [Google Scholar] [CrossRef]
- Zimm, B.H.; Stockmayer, W.H. The dimensions of chain molecules containing branches and rings. J. Chem. Phys. 1949, 17, 1301–1314. [Google Scholar] [CrossRef]
- Casassa, E.F. Some statistical properties of flexible ring polymers. J. Polym. Sci. Part A 1965, 3, 605. [Google Scholar] [CrossRef]
- Burchard, W.; Schmidt, M. Static and Dynamic Structure Factors Calculated for Flexible Ring Macromolecules. Polymer 1980, 21, 745–749. [Google Scholar] [CrossRef]
- Cates, M.E.; Deutsch, J.M. Conjectures on the Statistics of Ring Polymers. J. Phys. 1986, 47, 2121–2128. [Google Scholar] [CrossRef]
- Klein, J. Dynamics of Entangled Linear, Branched, and Cyclic Polymers. Macromolecules 1986, 19, 105–118. [Google Scholar] [CrossRef]
- Brown, S.; Szamel, G. Computer simulation study of the structure and dynamics of ring polymers. J. Chem. Phys. 1998, 109, 6184–6192. [Google Scholar] [CrossRef]
- Brown, S.; Szamel, G. Structure and dynamics of ring polymers. J. Chem. Phys. 1998, 108, 4705–4708. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, J.M. Equilibrium size of large ring molecules. Phys. Rev. E 1999, 59, R2539–R2541. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.; Jeong, C.; Winkler, R.G.; Lacevic, N.; Gee, R.H.; Yoon, D.Y. Chain Dynamics of Ring and Linear Polyethylene Melts from Molecular Dynamics Simulations. Macromolecules 2011, 44, 2311–2315. [Google Scholar] [CrossRef]
- Hur, K.; Winkler, R.G.; Yoon, D.Y. Comparison of ring and linear polyethylene from molecular dynamics simulations. Macromolecules 2006, 39, 3975–3977. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Takano, A.; Matsushita, Y. Topological effect in ring polymers investigated with Monte Carlo simulation. J. Chem. Phys. 2008, 129, 034903. [Google Scholar] [CrossRef]
- Suzuki, J.; Takano, A.; Deguchi, T.; Matsushita, Y. Dimension of ring polymers in bulk studied by Monte-Carlo simulation and self-consistent theory. J. Chem. Phys. 2009, 131, 144902. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Wittmer, J.P.; Cates, M.E. Topological effects in ring polymers: A computer simulation study. Phys. Rev. E 1996, 53, 5063–5074. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, G.; Tsalikis, D.; Mavrantzas, V. Microscopic Dynamics and Topology of Polymer Rings Immersed in a Host Matrix of Longer Linear Polymers: Results from a Detailed Molecular Dynamics Simulation Study and Comparison with Experimental Data. Polymers 2016, 8, 283. [Google Scholar] [CrossRef]
- Halverson, J.D.; Kremer, K.; Grosberg, A.Y. Comparing the results of lattice and off-lattice simulations for the melt of nonconcatenated rings. J. Phys. A Math. Theor. 2013, 46, 065002. [Google Scholar] [CrossRef]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. I. Statics. J. Chem. Phys. 2011, 134, 204904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. II. Dynamics. J. Chem. Phys. 2011, 134, 204905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsalikis, D.G.; Mavrantzas, V.G. Size and Diffusivity of Polymer Rings in Linear Polymer Matrices: The Key Role of Threading Events. Macromolecules 2020, 53, 803–820. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University: Oxford, UK, 1986. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Graessley, W.W. Polymeric Liquids & Networks: Structure and Properties; Taylor & Frances Group: London, UK; New York, NY, USA, 2008. [Google Scholar]
- Kapnistos, M.; Lang, M.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Richter, D.; Cho, D.; Chang, T.; Rubinstein, M. Unexpected power-law stress relaxation of entangled ring polymers. Nat. Mater. 2008, 7, 997–1002. [Google Scholar] [CrossRef]
- Higgins, J.S.; Benoit, H.C. Polymers and Neutron Scattering; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Richter, D.; Ewen, B. Neutron Spin-Echo Investigations on the Dynamics of Polymer Systems. In Relaxation in Polymers; Pietralla, M., Pechhold, W., Eds.; Springer-Verlag: New York, NY, USA, 1989; Volume 80, pp. 53–62. [Google Scholar]
- Hammouda, B. SANS from Polymers—Review of the Recent Literature. J. Macromol. Sci. Part C Polym. Rev. 2010, 50, 14–39. [Google Scholar] [CrossRef]
- Richter, D.; Goossen, S.; Wischnewski, A. Celebrating Soft Matter’s 10th Anniversary: Topology matters: Structure and dynamics of ring polymers. Soft Matter 2015, 11, 8535–8549. [Google Scholar] [CrossRef]
- Semlyen, J.A. Introduction: Cyclic Polymers—The First 40 Years. In Cyclic Polymers; Semlyen, J.A., Ed.; Springer: Dordrecht, The Netherlands, 2002; pp. 1–46. [Google Scholar]
- Higgins, J.S.; Dodgson, K.; Semlyen, J.A. Studies of Cyclic And Linear Poly(dimethyl siloxanes): 3. Neutron-Scattering Measurements of the Dimensions of Ring and Chain Polymers. Polymer 1979, 20, 553–558. [Google Scholar] [CrossRef]
- Edwards, C.J.C.; Richards, R.W.; Stepto, R.F.T.; Dodgson, K.; Higgins, J.S.; Semlyen, J.A. Studies of Cyclic And Linear Poly(dimethyl siloxanes). 14. Particle Scattering Functions. Polymer 1984, 25, 365–368. [Google Scholar] [CrossRef]
- Roovers, J.; Toporowski, P.M. Synthesis of High Molecular-Weight Ring Polystyrenes. Macromolecules 1983, 16, 843–849. [Google Scholar] [CrossRef]
- Roovers, J. Melt Properties of Ring Polystyrenes. Macromolecules 1985, 18, 1359–1361. [Google Scholar] [CrossRef]
- Takano, A.; Ohta, Y.; Masuoka, K.; Matsubara, K.; Nakano, T.; Hieno, A.; Itakura, M.; Takahashi, K.; Kinugasa, S.; Kawaguchi, D.; et al. Radii of Gyration of Ring-Shaped Polystyrenes with High Purity in Dilute Solutions. Macromolecules 2012, 45, 369–373. [Google Scholar] [CrossRef]
- Takano, A.; Kushida, Y.; Ohta, Y.; Masuoka, K.; Matsushita, Y. The second virial coefficients of highly-purified ring polystyrenes in cyclohexane. Polymer 2009, 50, 1300–1303. [Google Scholar] [CrossRef]
- Arrighi, V.; Gagliardi, S.; Dagger, A.C.; Semlyen, J.A.; Higgins, J.S.; Shenton, M.J. Conformation of cyclics and linear chain polymers in bulk by SANS. Macromolecules 2004, 37, 8057–8065. [Google Scholar] [CrossRef]
- Gagliardi, S.; Arrighi, V.; Ferguson, R.; Dagger, A.C.; Semlyen, J.A.; Higgins, J.S. On the difference in scattering behavior of cyclic and linear polymers in bulk. J. Chem. Phys. 2005, 122, 064904. [Google Scholar] [CrossRef] [Green Version]
- Bras, A.R.; Pasquino, R.; Koukoulas, T.; Tsolou, G.; Holderer, O.; Radulescu, A.; Allgaier, J.; Mavrantzas, V.G.; Pyckhout-Hintzen, W.; Wischnewski, A.; et al. Structure and dynamics of polymer rings by neutron scattering: Breakdown of the Rouse model. Soft Matter 2011, 7, 11169–11176. [Google Scholar] [CrossRef] [Green Version]
- Beaucage, G.; Kulkarni, A.S. Dimensional Description of Cyclic Macromolecules. Macromolecules 2010, 43, 532–537. [Google Scholar] [CrossRef]
- Griffiths, P.C. Newutron Scattering and Nuclear Magnetic Resonance Investigations of Cyclic Polymers. In Cyclic Polymers; Semlyen, J.A., Ed.; Springer: Dordrecht, The Netherlands, 2002. [Google Scholar]
- Dodgson, K.; Sympson, D.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethyl siloxanes): 2. Preparative Gel-Permeation Chromatography. Polymer 1978, 19, 1285–1289. [Google Scholar] [CrossRef]
- Beevers, M.S.; Mumby, S.J.; Clarson, S.J.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethyl siloxanes): 13. Static Dielectric Measurements and Dipole-moments. Polymer 1983, 24, 1565–1570. [Google Scholar] [CrossRef]
- Hild, G.; Strazielle, C.; Rempp, P. Cyclic Macromolecules—Synthesis and Characterization of Ring-Shaped Polystyrenes. Eur. Polym. J. 1983, 19, 721–727. [Google Scholar] [CrossRef]
- Duval, M.; Lutz, P.; Strazielle, C. Hydrodynamic Dimensions of Ring-Shaped Macromolecules in a Good Solvent. Makromol. Chem. Rapid Commun. 1985, 6, 71–76. [Google Scholar] [CrossRef]
- Roovers, J. Viscoelastic Properties of Polybutadiene Rings. Macromolecules 1988, 21, 1517–1521. [Google Scholar] [CrossRef]
- Nam, S.; Leisen, J.; Breedveld, V.; Beckham, H.W. Dynamics of unentangled cyclic and linear poly(oxyethylene) melts. Polymer 2008, 49, 5467–5473. [Google Scholar] [CrossRef]
- Ragnetti, M.; Geiser, D.; Hocker, H.; Oberthur, R.C. Small-Angle Neutron-Scattering (SANS) of Cyclic and Linear Polystyrene in Toluene. Makromol. Chem. Macromol. Chem. Phys. 1985, 186, 1701–1709. [Google Scholar] [CrossRef]
- Lee, H.C.; Lee, H.; Lee, W.; Chang, T.; Roovers, J. Fractionation of Cyclic Polystyrene from Linear Precursor by HPLC at the Chromatographic Critical Condition. Macromolecules 2000, 33, 8119–8121. [Google Scholar] [CrossRef]
- Gartner, T.E.; Haque, F.M.; Gomi, A.M.; Grayson, S.M.; Hore, M.J.A.; Jayaraman, A. Scaling Exponent and Effective Interactions in Linear and Cyclic Polymer Solutions: Theory, Simulations, and Experiments. Macromolecules 2019, 52, 4579–4589. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Milner, S.T.; Graessley, W.W. Packing Length Influence in Linear Polymer Melts on the Entanglement, Critical, and Reptation Molecular Weights. Macromolecules 1999, 32, 6847–6851. [Google Scholar] [CrossRef]
- Kramers, H.A. The Behavior of Macromolecules in Inhomogeneous Flow. J. Chem. Phys. 1946, 14, 415–424. [Google Scholar] [CrossRef]
- Ishizu, K.; Kanno, H. Novel synthesis and characterization of cyclic polystyrenes. Polymer 1996, 37, 1487–1492. [Google Scholar] [CrossRef]
- Roovers, J. Dilute-solution Properties of Ring Polystyrenes. J. Polym. Sci. Part B Polym. Phys. 1985, 23, 1117–1126. [Google Scholar] [CrossRef]
- Grosberg, A.Y. Critical Exponents for Random Knots. Phys. Rev. Lett. 2000, 85, 3858–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobay, A.; Dubochet, J.; Millett, K.; Sottas, P.E.; Stasiak, A. Scaling behavior of random knots. Proc. Natl. Acad. Sci. USA 2003, 100, 5611–5615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, H.; Yao, A.; Tsukahara, H.; Deguchi, T.; Furuta, K.; Inami, T. Average size of random polygons with fixed knot topology. Phys. Rev. E 2003, 68, 011102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, N.T.; Grosberg, A.Y. Limits of analogy between self-avoidance and topology-driven swelling of polymer loops. Phys. Rev. E 2005, 72, 061803. [Google Scholar] [CrossRef] [Green Version]
- Goossen, S.; Bras, A.R.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D.; Rubinstein, M.; Roovers, J.; Lutz, P.J.; Jeong, Y.; Chang, T.; et al. Influence of the Solvent Quality on Ring Polymer Dimensions. Macromolecules 2015, 48, 1598–1605. [Google Scholar] [CrossRef] [Green Version]
- Lutz, P.; McKenna, G.B.; Rempp, P.; Strazielle, C. Solution Properties of Ring-Shaped Polystyrenes. Makromol. Chem. Rapid Commun. 1986, 7, 599–605. [Google Scholar] [CrossRef]
- Pakula, T.; Geyler, S. Cooperative Relaxations in Condensed Macromolecular Systems. 3. Computer-Simulated Melts of Cyclic Polymers. Macromolecules 1988, 21, 1665–1670. [Google Scholar] [CrossRef]
- Khokhlov, A.R.; Nechaev, S.K. Topologically driven compatibility enhancement in the mixtures of rings and linear chains. J. Phys. II 1996, 6, 1547–1555. [Google Scholar] [CrossRef]
- Brown, S.; Lenczycki, T.; Szamel, G. Influence of topological constraints on the statics and dynamics of ring polymers. Phys. Rev. E 2001, 63, 052801. [Google Scholar] [CrossRef] [Green Version]
- Halverson, J.D.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Rheology of Ring Polymer Melts: From Linear Contaminants to Ring-Linear Blends. Phys. Rev. Lett. 2012, 108, 038301. [Google Scholar] [CrossRef] [Green Version]
- Iyer, B.V.S.; Lele, A.K.; Shanbhag, S. What is the size of a ring polymer in a ring-linear blend? Macromolecules 2007, 40, 5995–6000. [Google Scholar] [CrossRef]
- Subramanian, G.; Shanbhag, S. Conformational properties of blends of cyclic and linear polymer melts. Phys. Rev. E 2008, 77, 011801. [Google Scholar] [CrossRef] [PubMed]
- Wittmer, J.; Paul, W.; Binder, K. Rouse and Reptation Dynamics at Finite Temperatures—A Monte-Carlo Simulation. Macromolecules 1992, 25, 7211–7219. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Binder, K. Collective Diffusion, Nucleation, and Spinodal Decomposition in Polymer Mixtures. J. Chem. Phys. 1983, 79, 6387–6409. [Google Scholar] [CrossRef]
- Dagger, A.C.; Semlyen, J.A. Studies of cyclic and linear poly(dimethyl siloxanes): 33. Preparation and characterization of per-deuterated linears and cyclics. Polymer 1998, 39, 2621–2627. [Google Scholar] [CrossRef]
- Dagger, A.C.; Semlyen, J.A. Studies of cyclic and linear poly(dimethyl siloxanes): 34—Preparation, fractionation and characterisation of the first per-deuterated macrocyclic poly(dimethyl siloxanes). Polymer 1999, 40, 3243–3245. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Koukoulas, T.; Mavrantzas, V.G.; Pasquino, R.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Wischnewski, A.; Monkenbusch, M.; Richter, D. Microscopic Structure, Conformation, and Dynamics of Ring and Linear Poly(ethylene oxide) Melts from Detailed Atomistic Molecular Dynamics Simulations: Dependence on Chain Length and Direct Comparison with Experimental Data. Macromolecules 2017, 50, 2565–2584. [Google Scholar] [CrossRef]
- Bras, A.R.; Goossen, S.; Krutyeva, M.; Radulescu, A.; Farago, B.; Allgaier, J.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Compact structure and non-Gaussian dynamics of ring polymer melts. Soft Matter 2014, 10, 3649–3655. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Doi, Y.; Kinoshita, K.; Ohta, Y.; Takano, A.; Takahashi, Y.; Nagao, M.; Matsushita, Y. Conformations of Ring Polystyrenes in Bulk Studied by SANS. Macromolecules 2018, 51, 1539–1548. [Google Scholar] [CrossRef]
- Bensafi, A.; Maschke, U.; Benmouna, M. Cyclic polymers in good solvents. Polym. Int. 2000, 49, 175–183. [Google Scholar] [CrossRef]
- Obukhov, S.; Johner, A.; Baschnagel, J.; Meyer, H.; Wittmer, J.P. Melt of polymer rings: The decorated loop model. EPL Europhys. Lett. 2014, 105, 48005. [Google Scholar] [CrossRef]
- Pasquino, R.; Vasilakopoulos, T.C.; Jeong, Y.C.; Lee, H.; Rogers, S.; Sakellariou, G.; Allgaier, J.; Takano, A.; Bras, A.R.; Chang, T.; et al. Viscosity of Ring Polymer Melts. ACS Macro Lett. 2013, 2, 874–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, G.B.; Hostetter, B.J.; Hadjichristidis, N.; Fetters, L.J.; Plazek, D.J. A Study of the Linear Viscoelastic Properties of Cyclic Polystyrenes Using Creep and Recovery Measurements. Macromolecules 1989, 22, 1834–1852. [Google Scholar] [CrossRef]
- **ng, K.; Chatterjee, S.; Saito, T.; Gainaru, C.; Sokolov, A.P. Impact of Hydrogen Bonding on Dynamics of Hydroxyl-Terminated Polydimethylsiloxane. Macromolecules 2016, 49, 3138–3147. [Google Scholar] [CrossRef]
- Von Meerwall, E.; Ozisik, R.; Mattice, W.L.; Pfister, P.M. Self-diffusion of linear and cyclic alkanes, measured with pulsed-gradient spin-echo nuclear magnetic resonance. J. Chem. Phys. 2003, 118, 3867–3873. [Google Scholar] [CrossRef]
- Alatas, P.V.; Tsalikis, D.G.; Mavrantzas, V.G. Detailed Molecular Dynamics Simulation of the Structure and Self-Diffusion of Linear and Cyclic n-Alkanes in Melt and Blends. Macromol. Theor. Simul. 2016, 26, 1600049. [Google Scholar] [CrossRef]
- Rouse, P.E. A Theory of the Linear Viscoelastic Properties of Dilute Solutions of Coiling Polymers. J. Chem. Phys. 1953, 21, 1272–1280. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S. Dynamics of concentrated polymer systems. Part 1.—Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S. Dynamics of concentrated polymer systems. Part 2.—Molecular motion under flow. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1978, 74, 1802–1817. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S. Dynamics of concentrated polymer systems. Part 4.—Rheological properties. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1979, 75, 38–54. [Google Scholar] [CrossRef]
- Schleger, P.; Farago, B.; Lartigue, C.; Kollmar, A.; Richter, D. Clear Evidence of Reptation in Polyethylene from Neutron Spin-Echo Spectroscopy. Phys. Rev. Lett. 1998, 81, 124–127. [Google Scholar] [CrossRef]
- Klein, J.; Fletcher, D.; Fetters, L.J. Diffusional Behavior of Entangled Star Polymers. Nature 1983, 304, 526–527. [Google Scholar] [CrossRef]
- Allen, G.; Brier, P.N.; Goodyear, G.; Higgins, J.S. Motional broadening of the quasi-elastic peak in neutrons scattered from polymeric materials. Faraday Symp. Chem. S 1972, 6, 169–175. [Google Scholar] [CrossRef]
- Arrighi, V.; Gagliardi, S.; Ganazzoli, F.; Higgins, J.S.; Raffaini, G.; Tanchawanich, J.; Taylor, J.; Telling, M.T.F. Effect of Chain Length and Topological Constraints on Segmental Relaxation in Cyclic PDMS. Macromolecules 2018, 51, 7209–7223. [Google Scholar] [CrossRef]
- Higgins, J.S.; Ma, K.; Nicholson, L.K.; Hayter, J.B.; Dodgson, K.; Semlyen, J.A. Studies of Cyclic and Linear Poly(dimethyl siloxanes): 12. Observation of Diffusion Behavior by Quasielastic Neutron Scattering. Polymer 1983, 24, 793–799. [Google Scholar] [CrossRef]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics; Dover Publications: Mineola, NY, USA, 2000. [Google Scholar]
- Han, C.C.; Akcasu, A.Z. Dynamic light scattering of dilute polymer solutions in the nonasymptotic q-region. Macromolecules 1981, 14, 1080–1084. [Google Scholar] [CrossRef]
- Dodgson, K.; Higgins, J.S. Neutron Scattering from Cyclic Polymers. In Cyclic Polymers; Semlyen, J.A., Ed.; Springer: Dordrecht, The Netherlands, 1986. [Google Scholar]
- Tsolou, G.; Stratikis, N.; Baig, C.; Stephanou, P.S.; Mavrantzas, V.G. Melt Structure and Dynamics of Unentangled Polyethylene Rings Rouse Theory, Atomistic Molecular Dynamics Simulation, and Comparison with the Linear Analogues. Macromolecules 2010, 43, 10692–10713. [Google Scholar] [CrossRef]
- Goossen, S.; Bras, A.R.; Krutyeva, M.; Sharp, M.; Falus, P.; Feoktystov, A.; Gasser, U.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Molecular Scale Dynamics of Large Ring Polymers. Phys. Rev. Lett. 2014, 113, 168302. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.T.; Newhall, J.D. Stress Relaxation in Entangled Melts of Unlinked Ring Polymers. Phys. Rev. Lett. 2010, 105, 208302. [Google Scholar] [CrossRef]
- Allen, G.; Higgins, J.S.; Wright, C.J. Segmental Diffusion in Rubbers Studied by Neutron Quasi-Elastic Incoherent-Scattering. J. Chem. Soc. Faraday Trans. 2 1974, 2, 348–355. [Google Scholar] [CrossRef]
- Arrighi, V.; Gagliardi, S.; Zhang, C.H.; Ganazzoli, F.; Higgins, J.S.; Ocone, R.; Telling, M.T.F. A unified picture of the local dynamics of poly(dimethylsiloxane) across the melting point. Macromolecules 2003, 36, 8738–8748. [Google Scholar] [CrossRef]
- Arrighi, V.; Ganazzoli, F.; Zhang, C.H.; Gagliardi, S. New interpretation of local dynamics of poly(dimethyl siloxane) observed by quasielastic neutron scattering. Phys. Rev. Lett. 2003, 90, 058301. [Google Scholar] [CrossRef] [PubMed]
- Grapengeter, H.H.; Alefeld, B.; Kosfeld, R. An Investigation of Micro-Brownian Motions in Polydimethylsiloxane by Complementary Incoherent-Neutron-Scattering and Nmr Experiments Below Room-Temperature. Colloid Polym. Sci. 1987, 265, 226–233. [Google Scholar] [CrossRef]
- Kirst, K.U.; Kremer, F.; Pakula, T.; Hollingshurst, J. Molecular-Dynamics of Cyclic and Linear Poly(dimethylsiloxanes). Colloid Polym. Sci. 1994, 272, 1420–1429. [Google Scholar] [CrossRef]
- Roland, C.M.; Nagi, K.L. Segmental relaxation in poly(dimethylsiloxane). Macromolecules 1996, 29, 5747–5750. [Google Scholar] [CrossRef]
- Goodwin, A.A.; Beevers, M.S.; Clarson, S.J.; Semlyen, J.A. Cyclic polysiloxanes: 6. Dielectric relaxation of polymethylphenylsiloxanes. Polymer 1996, 37, 2597–2602. [Google Scholar] [CrossRef]
- Goodwin, A.A.; Beevers, M.S.; Clarson, S.J.; Semlyen, J.A. Studies of cyclic and linear polydimethylsiloxanes: 32. Dielectric relaxation investigations. Polymer 1996, 37, 2603–2607. [Google Scholar] [CrossRef]
- Smith, J.S.; Borodin, O.; Smith, G.D. A quantum chemistry based force field for poly(dimethylsiloxane). J. Phys. Chem. B 2004, 108, 20340–20350. [Google Scholar] [CrossRef]
- Ngai, K.L.; Colmenero, J.; Alegria, A.; Arbe, A. Interpretation of Anomalous Momentum-Transfer Dependences of Local Chain Motion of Polymers Observed by Quasi-Elastic Incoherent Neutron-Scattering Experiments. Macromolecules 1992, 25, 6727–6729. [Google Scholar] [CrossRef]
- Colmenero, J.; Alegria, A.; Alberdi, J.M.; Alvarez, F.; Frick, B. Dynamics of the Alpha-Relaxation of A Glass-Forming Polymeric System—Dielectric, Mechanical, NMR, and Neutron-Scattering Studies. Phys. Rev. B 1991, 44, 7321–7329. [Google Scholar] [CrossRef] [PubMed]
- Ganazzoli, F.; Raffaini, G.; Arrighi, V. The stretched-exponential approximation to the dynamic structure factor in non-entangled polymer melts. Phys. Chem. Chem. Phys. 2002, 4, 3734–3742. [Google Scholar] [CrossRef]
- Ragupathy, L.; Arrighi, V.; Cowie, J.M.G.; Ferguson, R.; McEwen, I.J.; Shenoy, S.L. Miscibility study of stereoregular poly(methyl methacrylate) blends. Experimental determination of phase diagrams and predictions. Macromolecules 2007, 40, 1667–1674. [Google Scholar] [CrossRef]
- Arrighi, V.; Higgins, J.S. Polymer blends. In Applications of Neutron Scattering to Soft Condensed Matter; Gabrys, B.J., Ed.; CRC Press: Boca Raton, FL, USA, 2000; pp. 227–266. [Google Scholar]
- Gooßen, S.; Krutyeva, M.; Sharp, M.; Feoktystov, A.; Allgaier, J.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Sensing Polymer Chain Dynamics through Ring Topology: A Neutron Spin Echo Study. Phys. Rev. Lett. 2015, 115, 148302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaue, T.; Nakajima, C.H. Miscibility phase diagram of ring-polymer blends: A topological effect. Phys. Rev. E 2016, 93, 042502. [Google Scholar] [CrossRef] [Green Version]
- Sakaue, T. Statistical physics of ring polymers based on topological volume concept. React. Funct. Polym. 2019, 134, 150–155. [Google Scholar] [CrossRef]
- Sakaue, T. Statistics and geometrical picture of ring polymer melts and solutions. Phys. Rev. E 2012, 85, 021806. [Google Scholar] [CrossRef] [Green Version]
- Sakaue, T. Ring Polymers in Melts and Solutions: Scaling and Crossover. Phys. Rev. Lett. 2011, 106, 167802. [Google Scholar] [CrossRef] [Green Version]
- Nachlis, W.L.; Bendler, J.T.; Kambour, R.P.; MacKnight, W.J. Topological Effects on Blend Miscibility. Macromolecules 1995, 28, 7869–7878. [Google Scholar] [CrossRef] [Green Version]
- Santore, M.M.; Han, C.C.; Mckenna, G.B. A Comparison of the Thermodynamic Stability and Phase-Separation Kinetics of Polymer Blends Containing Cyclic Chains of High-Molecular-Weight. Macromolecules 1992, 25, 3416–3423. [Google Scholar] [CrossRef]
- Kuo, C.M.; Clarson, S.J. An investigation of the thermodynamic interactions of oligomeric cyclic methylphenylsiloxanes in siloxane melts and blends. Polymer 2000, 41, 5993–6002. [Google Scholar] [CrossRef]
- Singla, S.; Beckham, H.W. Miscible Blends of Cyclic Poly(oxyethylene) in Linear Polystyrene. Macromolecules 2008, 41, 9784–9792. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Koukoulas, T.; Mavrantzas, V.G. Dynamic, conformational and topological properties of ring-linear poly(ethylene oxide) blends from molecular dynamics simulations. React. Funct. Polym. 2014, 80, 61–70. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Doi, Y.; Abdul Rahman, S.S.; Kim, E.; Kim, T.-H.; Takano, A.; Matsushita, Y. SANS Study of Ring Topology Effects on the Miscibility of Polymer Blends. Macromolecules 2018, 51, 1885–1893. [Google Scholar] [CrossRef]
- White, R.P.; Lipson, J.E.G.; Higgins, J.S. Effect of Deuterium Substitution on the Physical Properties of Polymer Melts and Blends. Macromolecules 2010, 43, 4287–4293. [Google Scholar] [CrossRef] [Green Version]
- Garas, G.E.; Kosmas, M.K. Chain correlations in linear and ring polymer blends. J. Chem. Phys. 1998, 108, 376–382. [Google Scholar] [CrossRef]
- Mckenna, G.B.; Plazek, D.J. The Viscosity of Blends of Linear and Cyclic Molecules of Similar Molecular Mass. Polym. Commun. 1986, 27, 304–306. [Google Scholar]
- Mckenna, G.B.; Plazek, D.J. Measurement of the Viscoelastic Properties of Blends of Linear and Cyclic Macromolecules. Abstr. Pap. Am. Chem. S 1987, 193, 161-POLY. [Google Scholar]
- Subramanian, G.; Shanbhag, S. Self-diffusion in binary blends of cyclic and linear polymers. Macromolecules 2008, 41, 7239–7242. [Google Scholar] [CrossRef] [Green Version]
- Kruteva, M.; Allgaier, J.; Richter, D. Direct Observation of Two Distinct Diffusive Modes for Polymer Rings in Linear Polymer Matrices by Pulsed Field Gradient (PFG) NMR. Macromolecules 2017, 50, 9482–9493. [Google Scholar] [CrossRef]
- Niedzwiedz, K.; Wischnewski, A.; Pyckhout-Hintzen, W.; Allgaier, J.; Richter, D.; Faraone, A. Chain Dynamics and Viscoelastic Properties of Poly(ethylene oxide). Macromolecules 2008, 41, 4866–4872. [Google Scholar] [CrossRef]
- Hammouda, B. Analysis of the Beaucage model. J. Appl. Crystallogr. 2010, 43, 1474–1478. [Google Scholar] [CrossRef]
- Hammouda, B. SANS from homogeneous polymer mixtures: A unified overview. In Polymer Characteristics; Springer: Berlin/Heidelberg, Germany, 1993; pp. 87–133. [Google Scholar]
- Doi, Y.; Matsumoto, A.; Inoue, T.; Iwamoto, T.; Takano, A.; Matsushita, Y.; Takahashi, Y.; Watanabe, H. Re-examination of terminal relaxation behavior of high-molecular-weight ring polystyrene melts. Rheol. Acta 2017, 56, 567–581. [Google Scholar] [CrossRef]
- Vlassopoulos, D.; Pasquino, R.; Snijkers, F. Progress in the Rheology of Cyclic Polymers. In Topological Polymer Chemistry: Progress of Cyclic Polymers in Syntheses, Properties and Functions; Tezuka, Y., Ed.; World Scientific Publishing: Singapore, 2014; pp. 291–316. [Google Scholar]
- Kawaguchi, D.; Masuoka, K.; Takano, A.; Tanaka, K.; Nagamura, T.; Torikai, N.; Dalgliesh, R.M.; Langridge, S.; Matsushita, Y. Comparison of interdiffusion behavior between cyclic and linear polystyrenes with high molecular weights. Macromolecules 2006, 39, 5180–5182. [Google Scholar] [CrossRef]
- Ozisik, R.; von Meerwall, E.D.; Mattice, W.L. Comparison of the diffusion coefficients of linear and cyclic alkanes. Polymer 2002, 43, 629–635. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Structure Repeat Unit | Mo (1)/g mol−1 | Me (2)/g mol−1 | Mc (3)/g mol−1 |
|---|---|---|---|---|
| PDMS |  | 74.015 | 12,000 | 24,500 |
| PEO |  | 44.05 | 2000 | 5870 |
| PS |  | 104.15 | 18,100 | 31,200 |
| PE |  | 28.05 | 1150 | 3480 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrighi, V.; Higgins, J.S. Local Effects of Ring Topology Observed in Polymer Conformation and Dynamics by Neutron Scattering—A Review. Polymers 2020, 12, 1884. https://doi.org/10.3390/polym12091884
Arrighi V, Higgins JS. Local Effects of Ring Topology Observed in Polymer Conformation and Dynamics by Neutron Scattering—A Review. Polymers. 2020; 12(9):1884. https://doi.org/10.3390/polym12091884
Chicago/Turabian StyleArrighi, Valeria, and Julia S. Higgins. 2020. "Local Effects of Ring Topology Observed in Polymer Conformation and Dynamics by Neutron Scattering—A Review" Polymers 12, no. 9: 1884. https://doi.org/10.3390/polym12091884