1. Introduction
Median age increases, especially in developed countries, are accompanied by an increase in the prevalence of several types of primary degenerative dementias, most of them associated with the elderly. However, differential diagnoses are difficult, since their etiopathogenic characteristics, as well as clinical behaviors, often overlap and become confusing.
Cerebrospinal fluid (CSF) is a clear, plasma-like fluid (an ultrafiltrate of plasma) that surrounds the brain and spinal cord. Recent studies have demonstrated the usefulness of CSF biomarkers in distinguishing between dementias in routine clinical settings, as well as indicating the presence of AD. Markers to consider in CSF are Aβ42 (sometimes normalized to the related peptide Aβ40), total tau (t-tau) protein, and phosphorylated tau (p-tau) protein. They make it possible to distinguish between AD, frontotemporal dementia, Lewy body dementia, Parkinson’s disease dementia, vascular dementia, and mixed dementia (AD and vascular) [
1].
Currently, it is possible to establish that AD is the most common cause of dementia, accounting for an estimated 60% to 80% of cases [
2], and the absence of an effective treatment, as well as the poor success in recent drug development efforts, is a growing health and social problem. For this reason, in recent years, intensive research has been carried out to understand the molecular mechanisms of AD and, based on this, to develop possible treatments.
AD pathogenesis is attributed to the loss of neuronal cells and the progressive atrophy of nervous tissue. However, neuronal loss shows important distinctions between different cell populations, the most affected being cholinergic neurons, which use acetylcholine (ACh) as a neurotransmitter. ACh is involved in various, higher cognitive processes, such as attention, learning, and memory. Moreover, cholinesterase inhibitors, such as donepezil, galantamine, and rivastigmine, have been commonly prescribed for AD for decades. However, although the inhibition of this esterase shows certain benefits, these drugs only provide temporary or incomplete symptomatic relief, and are not able to effectively slow the progression of AD [
3].
In the hippocampus, neuronal dysfunction can be derived from excitotoxicity, caused by consistently elevated glutamate levels, which cause excessive activation of the
N-methyl-
d-aspartate (NMDA) glutamate receptor, or from an increased sensitivity to glutamate, resulting in enhanced Ca
2+ flux reaching neurons, impaired neuronal homeostasis, and neurodegeneration [
4]. This justifies the use of memantine, a NMDA receptor antagonist for AD treatment.
Various histopathological and functional alterations that occur during AD progression are widely known. The most prominent include the formation of amyloid-β (Aβ) deposits and neurofibrillary tangles of p-tau protein in the neuronal framework, both used as markers in AD diagnosis, or the involvement of inflammatory processes.
The Aβ peptide comes from the aberrant processing of a neuronal transmembrane protein, known as amyloid precursor protein (APP). The key step in this pathway is the cleavage by β-secretase at the
N-terminus of Aβ, followed by another cleavage catalyzed by γ-secretase, resulting in the formation of Aβ oligomers (Aβ40 and Aβ42) that will polymerize, the neurotoxicity caused by Aβ42 being much greater [
5]. This aggregation entails, as a result, the blocking of ionic channels, the alteration of calcium homeostasis, an increase in mitochondrial oxidative stress, and a decrease in energy metabolism and glucose regulation, which contributes to the deterioration of neuronal health and, finally, to the death of neuronal cells [
6].
Another pathological feature of AD consists of the neurofibrillary changes represented by neurofibrillary tangles and neurofibrillary threads, which exhibit a stereotyped pattern of hierarchical progression initiated around the hippocampus. The hyperphosphorylation of tau, whose oligomers aggregate to form the neurofibrillary tangles, causes instability and collapse of the microtubules, loss of communication between neurons and, finally, neuronal apoptosis [
6]. Recent research data supports the theory that correlates the extent of neurofibrillary changes to the severity of dementia in AD [
7].
Therefore, aberrant proteins (abovementioned) are clearly associated with altered neurotransmission, neurodegeneration processes, and atrophy of brain structures. However, there are still numerous concerns about the causal sequence and interrelation of these events.
Considering the importance of the blood–brain barrier (BBB) to protect the central nervous system (CNS), the vascular hypothesis of the origin of AD has gained strength in the last two decades. It complements other existing theories, proposing that initial vascular damage precipitates AD. However, although the mechanisms linking BBB disruption and neurodegeneration provide an interesting basis for the search for new therapies for neurodegenerative diseases [
8], the precise mechanisms of BBB impairment are not fully understood.
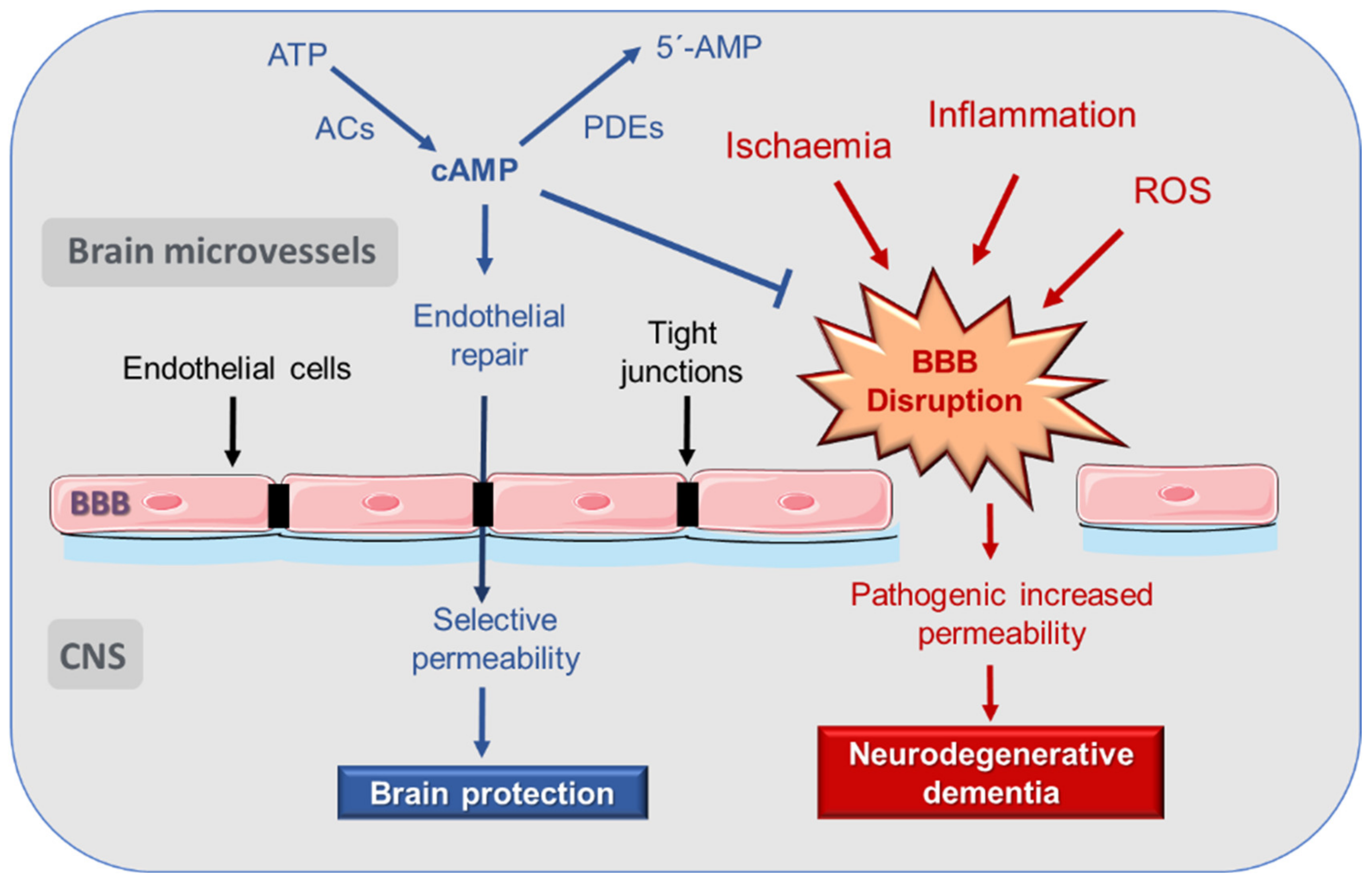
It is generally accepted that 3′,5′-cyclic adenosine monophosphate (cAMP) signaling participates in a prominent way in the regulation of endothelial permeability. An increase in cAMP concentrations improves the functionality of the endothelial barrier and increases tight junction (TJ) resistance in BBB [
9,
10]. Therefore, better knowledge of the local processes regulated by cAMP may be key when it comes to preserving the BBB function and preventing the progression of neurodegenerative diseases through pharmacological intervention.
This review proposes, for the first time, the possibility of modulating cAMP signaling pathways to preserve the cerebrovascular endothelial barrier function as a new strategy to prevent BBB hyperpermeability in neurodegenerative dementia, according to the vascular theory of AD. Our aim herein is to update knowledge on the role of cAMP and its complex and highly compartmentalized signaling in BBB disruption and propose new therapeutic targets for AD treatment.
2. The Vascular Hypothesis of Alzheimer’s Disease (AD)
In the search for new hypotheses that seek to explain the origins of neurodegenerative dementias, we considered that, in the elderly, vascular disorders make the brain more vulnerable, intensifying the symptoms of neurodegenerative disease.
From the observation that AD patients present reduced cerebral blood flow, cerebral oxygen consumption, and glucose metabolism, proportional to the severity of the disease, the vascular hypothesis has emerged as an explanation for the origin of AD [
11]. In accordance with this hypothesis, vascular and circulatory alterations in the brain precede the appearance of clinical symptoms by years or decades [
12]. In fact, post-mortem studies suggest that more than half of AD patients had cerebrovascular abnormalities [
13]. Moreover, clinical, imaging, neuropathological, and epidemiological evidence have been accumulated, confirming a key role of cerebrovascular disease in AD and other age-associated dementias [
14].
Currently, vascular dysfunction is considered a key factor in the development of AD. However, it is not yet clear whether vascular abnormalities precede or follow AD pathology, and the role of microcirculation in neurodegenerative disorders has not been sufficiently considered until now. This has probably contributed significantly to the failure of clinical trials, with various neuroprotective drugs.
A systematic review of the literature showed that a variety of disorders affecting small arteries and microvessels of the brain are associated with AD and may be related to Aβ/tau pathology [
15]. Thus, a large body of evidence has been presented to show that vascular disorders are involved in the pathogenesis of AD. A study demonstrated the prevalence of large infarcts, multiple lacunae and microinfarctions, hemorrhages, atherosclerosis, and arteriolosclerosis in 80% of patients diagnosed with AD [
16]. In addition, several studies have shown that vascular risk factors, such as dyslipidemia and hypertension, contribute to the development of AD [
17]. Although there have been few advances (in terms of therapy) in this regard, long-term treatment with the anticoagulant dabigatran improves cerebral perfusion and BBB function, preserving cognitive function and demonstrating the potential of a cardiovascular treatment to delay AD progression [
18].
Even in patients suffering from the genetic form of AD, many functional studies support the vascular hypothesis that drives the AD phenotype. A degenerative response of endothelial cells and pericytes mediated by cyclophilin A and nuclear factor kappa B (NF-κB) were observed in mice expressing human APOE4, the main genetic risk factor for AD [
19]. This response is also seen in APOE4-bearing humans. Another fact that supports the idea that the first changes in the BBB could play a main role in the initiation of AD is that the heterozygous deficiency of glucose transporter 1 (GLUT1) accelerates the degenerative changes in the endothelium in the APPsw mouse model for AD [
20].
Vascular dysfunction has a complex etiology (e.g., it is related to an increase in oxidative stress and inflammation). Mediators that are released have negative effects on the brain’s microvascular endothelial cells forming the BBB, as well as on other components of the neurovascular unit (NVU), such as astrocytes or microglia, causing an abnormal increase in permeability [
8]. Vascular damage also influences the Aβ-mediated neurodegeneration pattern, as the resulting BBB dysfunction leads to poor clearance of this peptide, contributing to its accumulation in the brain and the subsequent neurotoxic effects [
21].
The integrity of the NVU is necessary for the proper functioning of the brain, and damage at this level can lead to different diseases that affect the CNS. Currently, there is extensive evidence that NVU dysfunction contributes to AD [
22].
3. Disruption of Blood–Brain Barrier (BBB) in AD
The vascular hypothesis of AD suggest that the BBB plays a key role in the pathogenesis of Alzheimer’s disease. The BBB is a cell membrane that covers brain microvessels. It is composed of vascular endothelial cells of the microvasculature of the CNS, which communicate with other cells of the CNS, such as astrocytes, pericytes, and neurons, constituting the NVU [
23,
24].
Regarding the whole vascular system, the endothelium constitutes the innermost layer of all blood vessels, and one of its most prominent functions is the formation of a structural barrier that, through TJs, regulates the exchange of solutes between tissues and the blood, and the extravasation of multiple substances, platelets, and blood cells [
25,
26]. It also plays a crucial role in the control of multiple important functions, acting as an endocrine organ. Among its functions is the regulation of the regional blood flow, controlling vascular tone by releasing various vasodilator molecules, such as nitric oxide (NO), prostacyclin (PGI2), or the endothelium-derived hyperpolarizing factor (EDHF), and contracting factors, such as endothelins [
27,
28]. It also participates in the regulation of inflammation and angiogenesis [
29]. Therefore, it is not surprising that endothelial alterations are implicated in the pathogenesis and evolution of a wide spectrum of diseases [
25,
30,
31,
32].
Each NVU component plays an important role in maintaining the proper functioning of the BBB. Endothelial cells express multiple substrate-specific transport systems, which control the passage of essential molecules from the blood to the brain, as well as the transport of metabolic waste products from the interstitial fluid to the blood. In addition, endothelial cells are connected to each other by TJs, which link contiguous cells by multiple transmembrane proteins. This compact structure endows the endothelium with low permeability, which is reinforced by a covering formed by pericytes and astrocyte feet. Moreover, the pre-conditioning of human umbilical vein endothelial cells (HUVEC) with astrocyte-conditioned medium reduces the permeability to levels similar to that of the BBB [
33]. Microglia cells act as the first line of defense, while pericytes act by regulating angiogenesis, toxin elimination, brain flow, and entry of immune cells through the BBB. For their part, astrocytes guarantee brain metabolism, regulate synaptic transmission and plasticity, and prevent neuronal toxicity [
34,
35,
36].
The BBB allows the selective passage of nutrients and energy to the brain, which are essential for neuronal function and preventing the entry of neurotoxic substances from the peripheral circulation. Therefore, BBB has a crucial role in the maintenance of homeostasis in the CNS by limiting transport of toxic or harmful molecules, transport of nutrients, and removal of metabolites from the brain [
24,
28]. Transport through the endothelial cells is mainly mediated by the expression of receptors and transporters, such as the receptor for advanced glycosylation end products (RAGE), low-density lipoprotein receptor-related protein 1 (LRP-1), and P-glycoprotein (P-gp) [
37].
In certain pathological situations, the endothelium may undergo dysfunction, and its permeability may increase abnormally [
38]. Consequently, an endothelial dysfunction at the cerebral microvasculature may lead to a pathological fenestration of the BBB, with the subsequent abnormal increase of the barrier permeability and neuronal damage [
39].
It is generally accepted that BBB dysfunction occurs in AD, even before neurodegeneration and dementia. In fact, an increase in the normally low permeability of the BBB appears in the pathogenesis of various neurological diseases, and is present before the appearance of the first clinical symptoms [
23,
24].
The brains of AD patients were found to have lower expressions of various proteins involved in the formation of the TJs of the BBB endothelial cells, such as claudin-5, occludin, and zonula occludens-1 (ZO-1) [
40,
41]. Moreover, an increase in endothelial RAGE reactivity has been found in these patients, while the expression of LRP-1 and the function and expression of P-gp are reduced, decreasing the clearance of Aβ peptide from the brain [
37].
BBB disruption leads to the entry of blood constituents into the CNS, alters the clearance mechanism, and is associated with reduced cerebral flow. Recently, it was also shown that, at the endothelial level, cerebral blood flow can be regulated by GLUT1. GLUT1 is further required to maintain the integrity of BBB and proper brain capillary networks, as well as neuronal function. Its deficiency in endothelium initiates the vascular phenotype of AD, as shown by BBB breakdown. In GLUT1-deficient AD mice (Slc2a1+/− APPSw/0), reduced brain perfusion and diminished glucose uptake into the brain occur at 2 weeks of age. However, neuronal dysfunction, behavioral deficits, elevated Aβ levels, and behavioral and neurodegenerative changes take approximately 6 months to develop [
20]. Based on these (and other) results, BBB disruption is currently considered an early indicator of cognitive dysfunction and AD.
Consequently, the mechanisms that relate BBB disruption and neurodegeneration constitute an interesting basis in the search for new therapies for neurodegenerative diseases (
Figure 1).
Thus far, the precise mechanisms responsible for BBB impairment are not fully understood. Research has focused on the role played by both the pericytes and the endothelial cells of the brain microvasculature in BBB disruption. In post-mortem studies, it was observed that a degeneration of the pericytes occurs in regions that presented elevated fibrillar Aβ, which induces or worsens BBB disruption [
42]. Likewise, it has been experimentally shown that apolipoprotein E (APOE), a risk factor for AD, activates the pro-inflammatory cyclophilin A-nuclear factor-κM-matrix-metalloproteinase-9 pathway in pericytes, leading to BBB impairment [
19].
Regarding endothelial dysfunction, aging is an independent factor, even in the absence of other cardiovascular risk factors. Vascular inflammation occurs with aging [
43,
44] and can be reinforced by metabolic and cardiovascular diseases [
45,
46]. Thrombin is an important mediator that produces vascular inflammation. It can directly activate endothelial cells and promote the expression of pro-inflammatory proteins, such as intercellular adhesion molecule-1 (ICAM-1) and monocyte chemoattractant protein-1 (MCP-1), the release of angiopoietin-2, and the positive regulation of αVβ3 integrin [
47]. Inflammatory mediators interact with leukocytes and reduce vasodilation-modifying BBB permeability [
48].
There are several sources of reactive oxygen species (ROS), which are altered in AD, such as mitochondrial electron-transport chain, cyclooxygenases (COXs), lipoxygenases, cytochrome P450 reductases, xanthine oxidase, nitric oxide synthase (NOS), and Nox. ROS produce oxidative stress and are responsible for altering protein structure, DNA denaturation, and lipid peroxidation, and may act as messengers in redox-signaling systems. Superoxide, hydroxyl radical, and hydrogen peroxide are common ROS with deleterious effects on the vascular endothelium. Their concentrations depend on the balance between oxidases, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox enzymes) and superoxide dismutase (SOD). The impact of ROS on BBB function was demonstrated in SOD deficient mice, in which ischemia/reperfusion experiments demonstrated an enhanced endothelial permeability to large molecules [
49].
Claudin-5 expression is regulated by ROS production, increasing the leakage of solute, and modifying the BBB integrity [
50]. Similarly, occludin expression is reduced by AMP-activated protein kinase (AMPK) activation that enhances lipopolysaccharide (LPS)-impaired BBB functions through suppression of NADPH oxidase-derived ROS in mice [
51]. In addition to claudin and occludin, ROS can change ZO protein distribution. Exposure to hydrogen peroxide has been reported to lead to the redistribution of ZO-1 from the TJs to the cytosol, resulting in decreased transepithelial electric resistance (TEER) and increased BBB permeability [
52]. Therefore, ROS cause changes in various parameters that compromise the integrity of the BBB.
The BBB damage produced by ROS is also related to the activation of transcription factors that produce a proinflammatory response, with NF-κB being the main regulator. Activation of NF-κB by ROS can increase ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) expression [
53]. ICAM-1 can activate a Ca
2+ signaling pathway that can lead to cytoskeleton changes in microvascular endothelial cells of the brain, causing BBB damage [
54].
ROS overproduction also interferes with hypoxia-inducible factor 1α (HIF-1α) reducing its expression and activity. Thus, the stimulus to promote angiogenesis and the formation of new vessels will be restricted, leading to a vicious cycle of impaired capillary perfusion, hypoxia, and oxidative stress [
55].
It follows—from the above—that a highly selective BBB permeability is essential for the maintenance of healthy brain functioning. In this sense, it is known that the cAMP signaling pathway plays a very prominent role in the regulation of cerebrovascular endothelial permeability, as reviewed in the following sections.
4. cAMP Regulation of Endothelial and BBB Permeability
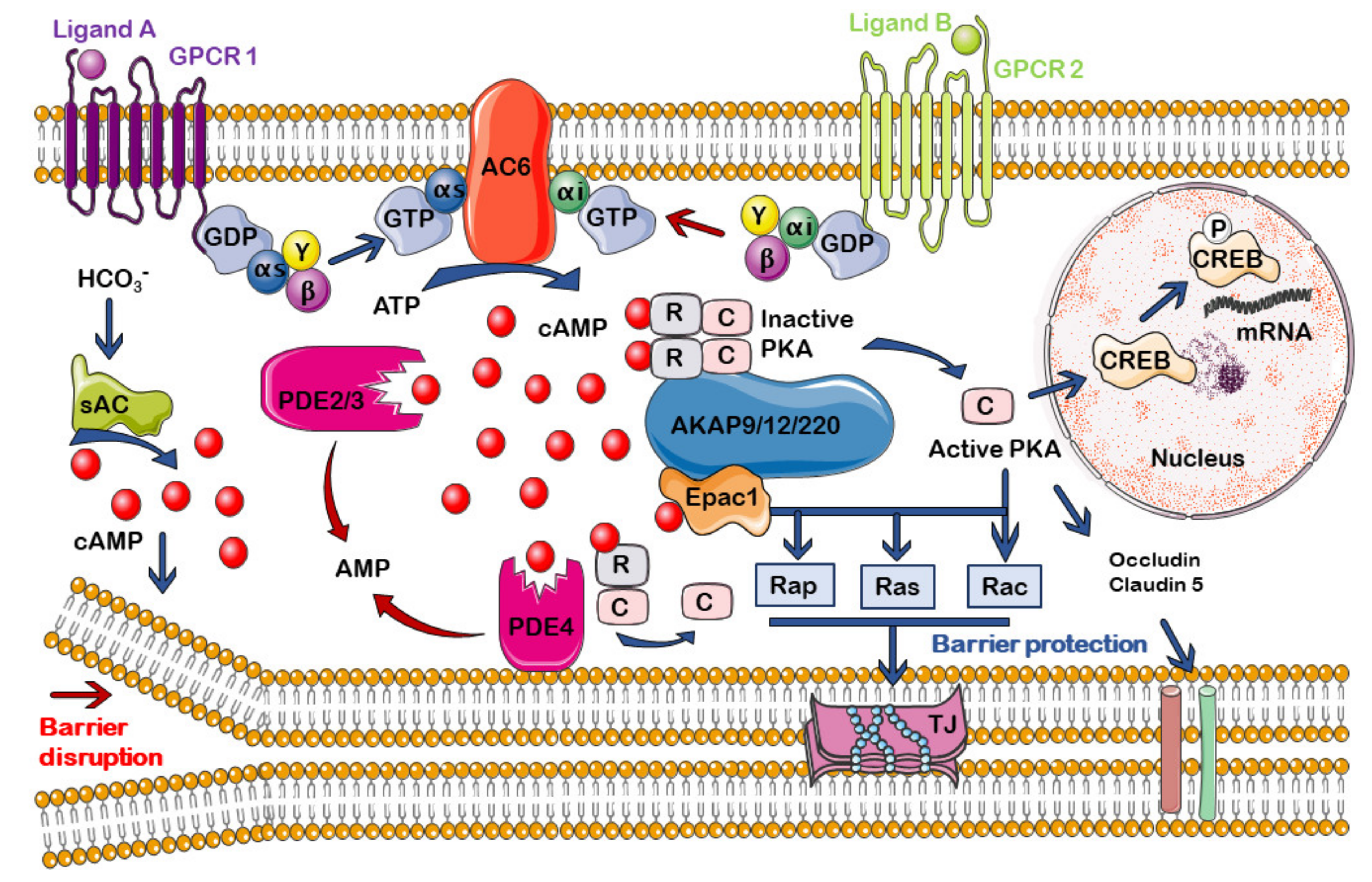
cAMP is a second universal messenger, generated when an extracellular first messenger (neurotransmitters, hormones, chemokines, lipid mediators, or drugs) binds to a G-protein coupled receptor (GPCR) associated with an AC enzyme that catalyzes the cyclization of adenosine triphosphate (ATP) to generate cAMP [
56,
57]. G proteins that regulate the intracellular concentration of cAMP are of two subtypes: Gs or Gi proteins, which stimulate or inhibit, respectively, the activity of adenylyl cyclases (ACs). cAMP intracellular concentration is also regulated by the family of phosphodiesterases (PDEs), which catalyze cAMP hydrolysis, ending the signaling pathway [
58].
cAMP participates in the regulation of many biological processes and cellular functions, such as metabolism, gene regulation, regulation of neurotransmitter synthesis, growth factors, and immune function [
59].
In the late 1990s, it was suggested that increases in cAMP concentration could be involved in a decrease in endothelial permeability [
60] (
Figure 1). By measuring microvascular hydraulic conductivity in mesentery of pithed frogs, it could be seen that cAMP decreases microvascular permeability in vivo by increasing the number of Tj strands between endothelial cells [
61]. Later studies served to demonstrate that cAMP signaling participates in a prominent way in the regulation of endothelial permeability, and a rise in its concentration enhances barrier functions [
10,
25,
62,
63,
64,
65]. This ability of cAMP to stabilize the barrier occurs in resting conditions and in the presence of barrier destabilizers [
66].
In regard to BBB—in 1991, a study using a new in vitro model of BBB, combining brain endothelial cells monolayers and astrocyte conditioned medium, demonstrated that cAMP-elevating agents increase TJ resistance. On the contrary, agents that decreased cAMP concentration or block activity reduced TJ resistance [
9]. A few years later, it was shown that various agents that increase cAMP (dibutyryl-cAMP, isoprenaline, and human α-calcitonin gene-related peptide) counteract permeability increases in pial venular capillaries, which are part of the BBB [
67].
The in vivo temporal regulation by cAMP of the BBB permeability to solutes was demonstrated in 2014 using multiphoton microscopy in rat brain parenchyma [
68]. It was shown that cAMP increase by stimulation of adenosine receptors induces gap junction coupling in human cerebral microvascular endothelial cells, an effect mediated by cyclic nucleotide-gated (CNG) channel activation and the subsequent increase in Ca
2+ influx [
69].
cAMP-activated pathways make up complex and highly compartmentalized signaling. Thus, it is no surprise that study results have also reported that, contrary to the widely accepted notion, an endothelial increase of cAMP could enhance vascular permeability, an effect that would be mediated by transcriptional small guanosine triphosphate hydrolase Ras-related protein (RRAS) suppression [
70].
In general, cAMP-mediated signaling can be very different, depending on the cell model. As an example, an increase in cAMP causes contraction of cardiac myocytes, but relaxation of vascular ones. In addition, there are also differences in the cAMP-elicited response depending on the microdomain in which its elevation occurs within the same cell type. Therefore, in the following section, we will address the current knowledge about cAMP compartmentalization, focusing on how it can affect the control of the permeability at the endothelium and the BBB.
7. cAMP Compartmentalization in the BBB and Aging
It is generally accepted that BBB dysfunction occurs during aging and there is increasing evidence of its close relationship with cognitive impairment [
170]. An abnormally high permeability of the BBB may constitute an early marker for predicting cognitive decline in AD progression. However, despite the prominent role that it plays in regulating endothelial permeability, knowledge about cAMP signaling pathways and compartmentalization at the BBB level is scarce, as can be seen in the previous sections.
Furthermore, there are almost no studies on cAMP signaling alterations during aging or neurodegenerative diseases. Consequently, at present, there are no studies on drugs that, acting on the different proteins involved in the compartmentalization of cAMP at the BBB, can provide a benefit in the treatment of AD. In any case, there are some studies that may serve as the basis for future studies on the influence of aging, on the regulation of cAMP signalosomes in the BBB.
It is important to note that most studies in this regard refer to the influence of age on the cAMP compartmentalization in neurons, not in the BBB, which is out of the scope of this review. With aging, there is a significant decrease in AC activity in both human and animal brains, leading to lower levels of cAMP, which may favor the development of neurodegenerative diseases. Thus, during aging, the local distribution of the cAMP signal in neurons is altered, a situation that can participate in the development of various diseases, including neurodegenerative processes, such as AD [
57]. However, there are no specific studies on cerebral microvasculature, demonstrating a similar decrease in AC activity or an alteration in its intracellular distribution that could lead to a progressive increase in the permeability of the BBB with aging.
There are few studies on the alteration of the expression of PDE in the brain during aging, which showed contradictory results [
57,
171]. Moreover, cilostazol treatment has been shown to exert beneficial long-term effects, reducing age-related cognitive decline in senescence accelerated mouse (SAMP8), a mouse model of cognitive aging. This effect is exerted by a mechanism related to the increase of cAMP in the brain and the protection of the integrity of the BBB [
172]. The role of PDEs in endothelial function in ischemic stroke, including regulation of permeability, has recently been reviewed [
135].
Similarly, the possible pathological alterations in the function of AKAPs during aging and their implication in cognitive deterioration have hardly been studied and there are no studies of this type at the endothelial barrier level [
13].
Finally, it was reported that cAMP-mediated relaxation in mice basilar arteries is mainly related to the activation of Epac versus PKA, and is diminished with endothelial and smooth muscle aging [
173]. However, as with other elements of the cAMP signaling pathway, most of the studies on the possible influence of Epac alterations in aged-related neurodegenerative diseases concerns its presence in the brain and there are no studies of its possible alteration at the BBB level [
171].
8. Conclusions and Perspectives
In the search for new, successful, therapeutic options to treat AD, the vascular hypothesis paves the way for potential strategies related to the improvement of blood flow at the cerebral microvascular and protection of the BBB, as highlighted in this review.
Preserving the permeability of this barrier—or even achieving a hypothetical reversal of existing pathological disruptions—could contribute toward slowing, or even stop**, the progression of neurodegenerative diseases, including primary degenerative dementias, such as AD.
The important role that cAMP plays in maintaining the endothelial barrier function is widely known. However, the compartmentalization of cAMP signaling has certainly been studied more in other barriers, such as pulmonary or retinal, or endothelial functioning, in general, using various animal or human cell models. As each barrier has its own peculiarities, both at the functional and molecular level, it seems necessary to gather more knowledge on the cAMP signaling pathway at the BBB, as well as on the spatial and temporal distribution of this signal at the endothelium that integrates this barrier.
The distribution of the different pools of cAMP has also been studied at the brain level. However, a therapeutic proposal based on the vascular hypothesis and the protection of the BBB for the treatment of AD involves the delivery of drugs to a target in the endothelium of the cerebral microvasculature. Therefore, it would not be necessary to use molecules that must cross the BBB towards the SNC.
The ubiquity of cAMP means that less selective drugs can exert actions in different areas of the body, which increases the risk of adverse effects. Therefore, the use of more selective drugs, targeting the various protein isoforms directly involved in regulating BBB permeability, represents a potentially more effective and safe strategy. In this sense, a new therapeutic approach to modify the course of the disease by reducing BBB permeability could include the enhancement of certain cAMP intracellular pools through pharmacological actions on different subtypes of ACs and PDEs, the selective activation or inhibition of PKA or Epac subtypes, or a strategy to induce the formation of certain molecular clusters by acting on anchor proteins, such as AKAP.
In conclusion, develo** a new strategy to treat AD, based on the ability of cAMP in protecting BBB integrity against disrupting pathogenic agents, makes it essential to gather more knowledge on i) the compartmentalization of cAMP-dependent processes that regulate BBB permeability; and ii) the development of molecules that can selectively modulate the agents involved in this compartmentalization and/or that can be directed in a more selective way towards the BBB.



{kind=link}
{kind=link}

