Computational Exploration of Functionalized Rhombellanes: Building Blocks and Double-Shell Structures


Abstract
:
1. Introduction
2. Methods
3. Results and Discussion
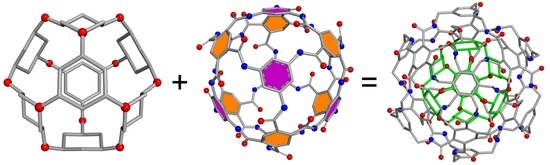
3.1. Structural Models
3.1.1. Core Building Blocks
3.1.2. Shell Building Blocks
3.1.3. Double-Shell Assemblies
3.2. Computational Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Canceill, J.; Lacombe, L.; Collet, A. A new cryptophane forming unusually stable inclusion complexes with neutral guests in a lipophilic solvent. J. Am. Chem. Soc. 1986, 108, 4230–4232. [Google Scholar] [CrossRef]
- Cram, D.J. Molecular container compounds. Nature 1992, 356, 29–36. [Google Scholar] [CrossRef]
- Sun, Q.F.; Murase, T.; Sato, S.; Fujita, M. A sphere-in-sphere complex by orthogonal self-assembly. Angew. Chem. Int. Ed. 2011, 50, 10318–10321. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, L.R.; Atwood, J.L. A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 1997, 389, 469–472. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, C.; Comotti, A.; Ward, M.D. Supramolecular archimedean cages assembled with 72 hydrogen bonds. Science 2011, 333, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Mück-Lichtenfeld, C.; Antony, J. Noncovalent interactions between graphene sheets and in multishell (hyper)fullerenes. J. Phys. Chem. C 2007, 111, 11199–11207. [Google Scholar] [CrossRef]
- Casella, G.; Bagno, A.; Saielli, G. Spectroscopic signatures of the carbon buckyonions C60@C180 and C60@C240: A dispersion–corrected DFT study. Phys. Chem. Chem. Phys. 2013, 15, 18030–18038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichierri, F. Hypercubane: DFT-based prediction of an Oh-symmetric double-shell hydrocarbon. Chem. Phys. Lett. 2014, 612, 198–202. [Google Scholar] [CrossRef]
- Pichierri, F. Substituent effects in cubane and hypercubane: A DFT and QTAIM study. Theor. Chem. Acc. 2017, 136, 114. [Google Scholar] [CrossRef]
- Diudea, M.V. Rhombellanic crystals and quasicrystals. Iran. J. Math. Chem. 2018, 9, 167–178. [Google Scholar]
- Diudea, M.V.; Nagy, C.L. Rhombellane space filling. J. Math. Chem. 2019, 57, 473–483. [Google Scholar] [CrossRef]
- Diudea, M.V.; Lungu, C.N.; Nagy, C.L. Cube-Rhombellane related structures: A drug perspective. Molecules 2018, 23, 2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeleń, P.; Szefler, B. The Immobilization of ChEMBL474807 Molecules Using Different Classes of Nanostructures. Symmetry 2019, 11, 980. [Google Scholar] [CrossRef] [Green Version]
- Czeleń, P.; Szefler, B. The immobilization of oxindole derivatives with use of cube rhombellane homeomorphs. Symmetry 2019, 11, 900. [Google Scholar] [CrossRef] [Green Version]
- Szefler, B.; Czeleń, P. Docking of cisplatin on fullerene derivatives and some cube rhombellane functionalized homeomorphs. Symmetry 2019, 11, 874. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Liu, Y.-L.; Chen, D.; Shang, P.; Yin, D.-C. A review of magnet systems for targeted drug delivery. J. Control. Release 2019, 302, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Meng, L.; Zheng, S. Density functional studies on a novel double–shell fullerene C20@C60. J. Mol. Struct. THEOCHEM 2005, 725, 17–21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Formula | Natoms | Symm. | Egap (eV) | Hf (a.u.) | Hf/N (kcal/mol) |
|---|---|---|---|---|---|---|
| 2a | C48O12H72 | 132 | Oh | 6.139 | −4.906 | −260.480 |
| 2b | C48O12H72 | 132 | Oh | 7.264 | −25.000 | −261.466 |
| 2c | C48O12H72 | 132 | Td | 6.448 | −25.142 | −262.948 |
| 2d | C48O12H72 | 132 | Oh | 6.843 | −25.041 | −261.895 |
| 2e | C48O12H24 | 84 | Oh | 5.899 | −20.120 | −210.428 |
| 2f | C48O12H48 | 108 | Td | 6.200 | −22.601 | −236.371 |
| 3a | C108N24O24H60 | 216 | T | 2.877 | −52.610 | −211.622 |
| 3b | C108O48H36 | 192 | T | 3.914 | −47.180 | −189.782 |
| 3c | C72N12O12H48 | 144 | T | 3.880 | −34.190 | −223.483 |
| 3d | C72O24H36 | 132 | T | 4.201 | −31.507 | −205.949 |
| 4a | C132N12O36H96 | 276 | T | 3.942 | −62.110 | −216.525 |
| 4b | C132O48H84 | 264 | T | 4.020 | −59.334 | −206.849 |
| 4c | C192N24O48H180 | 444 | T | 4.738 | −95.962 | −228.095 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, K.; Szefler, B.; Nagy, C.L. Computational Exploration of Functionalized Rhombellanes: Building Blocks and Double-Shell Structures. Symmetry 2020, 12, 343. https://doi.org/10.3390/sym12030343
Nagy K, Szefler B, Nagy CL. Computational Exploration of Functionalized Rhombellanes: Building Blocks and Double-Shell Structures. Symmetry. 2020; 12(3):343. https://doi.org/10.3390/sym12030343
Chicago/Turabian StyleNagy, Katalin, Beata Szefler, and Csaba L. Nagy. 2020. "Computational Exploration of Functionalized Rhombellanes: Building Blocks and Double-Shell Structures" Symmetry 12, no. 3: 343. https://doi.org/10.3390/sym12030343