1. Introduction
With 57,000 new cases and 46,000 deaths annually, pancreatic ductal adenocarcinoma (PDAC) is the 9th most frequent malignant disease in males and 8th most prevalent in females and the 4th most frequent cause of death in both genders [
1]. At the time of diagnosis, only a fraction of patients are amenable to surgical resection of the tumor. However, even patients who undergo complete surgical resection are at a high risk of either local or systemic recurrence [
2].
In order to study the molecular mechanisms of pathogenesis and metastasis of pancreatic cancer, several genetically induced mouse models (GEMMs) have been established in recent decades [
3,
4,
5,
6]. The most prominent and best-characterized model is the LSL-
KrasG12D, LSL-
Trp53R172H,
Pdx1-
Cre (KPC) mouse model [
6].
It is now well established that circulating tumor cells (CTCs) are an integral part of the metastatic cascade in malignant disease [
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18]. CTCs are shed from the primary tumor, survive in circulation, and ultimately colonize distant organs where they establish clinically overt metastases [
9,
16]. However, while thousands or millions of CTCs are shed into the bloodstream over time, the number of metastases is several orders of magnitude lower [
19]. Therefore, it can be assumed that only a small fraction of CTCs are actually tumorigenic and thus clinically relevant. In order to achieve the invasive phenotype required to leave the primary tumor bulk and enter circulation, CTCs from epithelial tumors undergo a process called epithelial–mesenchymal transition (EMT). During this process, the cells acquire mesenchymal properties (e.g., migratory capability) while downregulating epithelial traits [
20,
21]. This process is reverted during mesenchymal-epithelial transition (MET). As both EMT and MET dynamically fluctuate, CTCs in circulation exhibit a high degree of plasticity and represent a heterogeneous population consisting of epithelial, mesenchymal, and intermediary cells [
20,
22].
In a previous study, CTCs from the GEMM (LSL-
KrasG12D, LSL-
Trp53flox/flox or flox/+,
Pdx1-
Cre) were isolated and submitted to single-cell RNA sequencing. The raw data of this experiment was released to the Gene Expression Omnibus (GEO) database, and it is available for analysis [
23]. In previous studies from our own group, we were able to demonstrate that, apart from immune evasive capabilities, CTCs also possess stem cell properties [
13,
17]. The main goal of this study was therefore to investigate the role of stem cell properties in murine CTCs of PDAC and to further characterize CTCs with stem cell properties.
3. Discussion
The aim of this study was to identify and characterize pancreatic CTCs with stem-like characteristics and explore potential underlying mechanisms through integrated bioinformatics analysis.
Numerous molecular markers, including
Cd24, Cd44, Prom1 (
Cd133), and other genes, have been applied in order to define pancreatic cancer stem cells (CSCs) [
30,
37,
38]. The CTC subgroup identified in this study as CTC-S demonstrated heterogeneous expression of stemness and pluripotency markers in comparison to CTC-N, with some markers showing increased expression (
Aldh1a2 and
Klf4), some with reduced levels (
Abcg2, Cd44, Cxcr4, Nanog and
Sox2) and others with no significant change in expression (
Aldh1a1, Cd24a, Met and
Prom1). The mutual exclusivity phenomenon may be due to the heterogeneous nature of stem-like cells and stemness markers [
39]. Similar results have been published for breast cancer, where
CD44high /
CD24low defines a cluster of tumor cells with stem-like features [
40]. This mutual exclusivity is confirmed in the here-presented results.
The invasive capabilities (e.g., migration and invasion) of tumor cells are a prerequisite for successful initiation and completion of metastasis. Accumulating evidence suggests that these features can be acquired through epithelial–mesenchymal transition (EMT), which is considered as a hallmark of tumor cell dissemination [
31,
41]. The CTC-S subpopulation exhibits a mixed epithelial/mesenchymal phenotype, while the CTC-N group exhibits neither epithelial nor mesenchymal markers. This is consistent with previous studies relating EMT to stemness [
42,
43]. Furthermore, CTCs with partial EMT phenotype or stem-like features may predict unfavorable survival in cancers independently [
44].
Epithelial markers (most prominently
EpCAM) have been widely used as identification and isolation markers of CTCs, and the prognostic value of
EpCAM-positive CTCs is evident [
15,
45,
46,
47,
48], but previous data have pointed towards a relative down-regulation of epithelial markers on CTCs [
13]. The here-presented results draw a more complex image of
EpCAM expression in CTCs—it can be speculated that
EpCAM expression on CTCs is a continuum from
EpCAM-negative to highly
EpCAM-positive cells. While, in this work,
EpCAM seems to be relatively overexpressed on cells with presumably higher metastatic potential, more research on the role of
EpCAM on CTCs is clearly required.
In theory, the loss of cell adhesion molecules during EMT should also coincide with the down-regulation of genes associated with adherens junctions. While both
Cdh1 and
Cdh2 (generally considered a mesenchymal marker) are downregulated in the CTC-S subgroup, most of the genes in the adherens junction pathway are up-regulated in comparison to CTC-N, resulting in a mixed epithelial/mesenchymal phenotype of CTC-S. This may represent a transitory state allowing CTC-S to undergo functional adaptation during the metastatic cascade [
49]. Additionally, this overexpression of adherens junctions may enable CTC-S to bind to endothelial cells prior to extravasation.
Intercellular adhesion has been demonstrated to be crucial for successful metastasis of epithelial cancers [
50]. Among the different types of intercellular adhesions, cell–cell adherens junctions are the most common and contribute to cell polarity, tissue architecture maintenance, cell movement limitation, and proliferation [
50]. Strong adherens junctions are mediated by the cadherin–catenin complex [
51,
52,
53]. The here-presented data show that the CTC-S cells express low levels of E-cadherin (
Cdh1), while the other genes from the adherens junction pathway were up-regulated, such as
Rhoa and
Cdc42, both of which are necessary for adherens junction maintenance [
54,
55]. We hypothesized that CTC-S cells are in the process of forming the adherens junction through actin cytoskeleton remodeling [
56].
Besides, another critical component of adherens junctions, plakoglobin (also known as γ-catenin), was found to be necessary for the formation of CTC clusters and further contributed to the metastatic cascade [
57,
58]. In summary, adherens junctions are critical to the formation of CTC clusters, which can form tumor-microemboli, ultimately outgrowing to overt metastases [
57]. These cellular aggregates have been detected in 81% of PDAC patients with unfavorable OS and PFS [
59]. However, it must be taken into account that all 75 pancreatic CTCs analyzed in this study were isolated individually, which means there are no data derived from CTC clusters in this dataset; the validity of the here-presented data concerning CTC clusters is therefore limited.
Numerous studies have explored the
Wnt signaling pathway, mainly due to the fact that canonical
Wnt signaling regulates, stabilizes, and promotes the accumulation of the β-catenin by inhibiting its degradation [
60,
61]. A few studies have demonstrated that
Wnt/β-catenin signaling plays a crucial role in the plasticity of stem cells [
62,
63]. Furthermore,
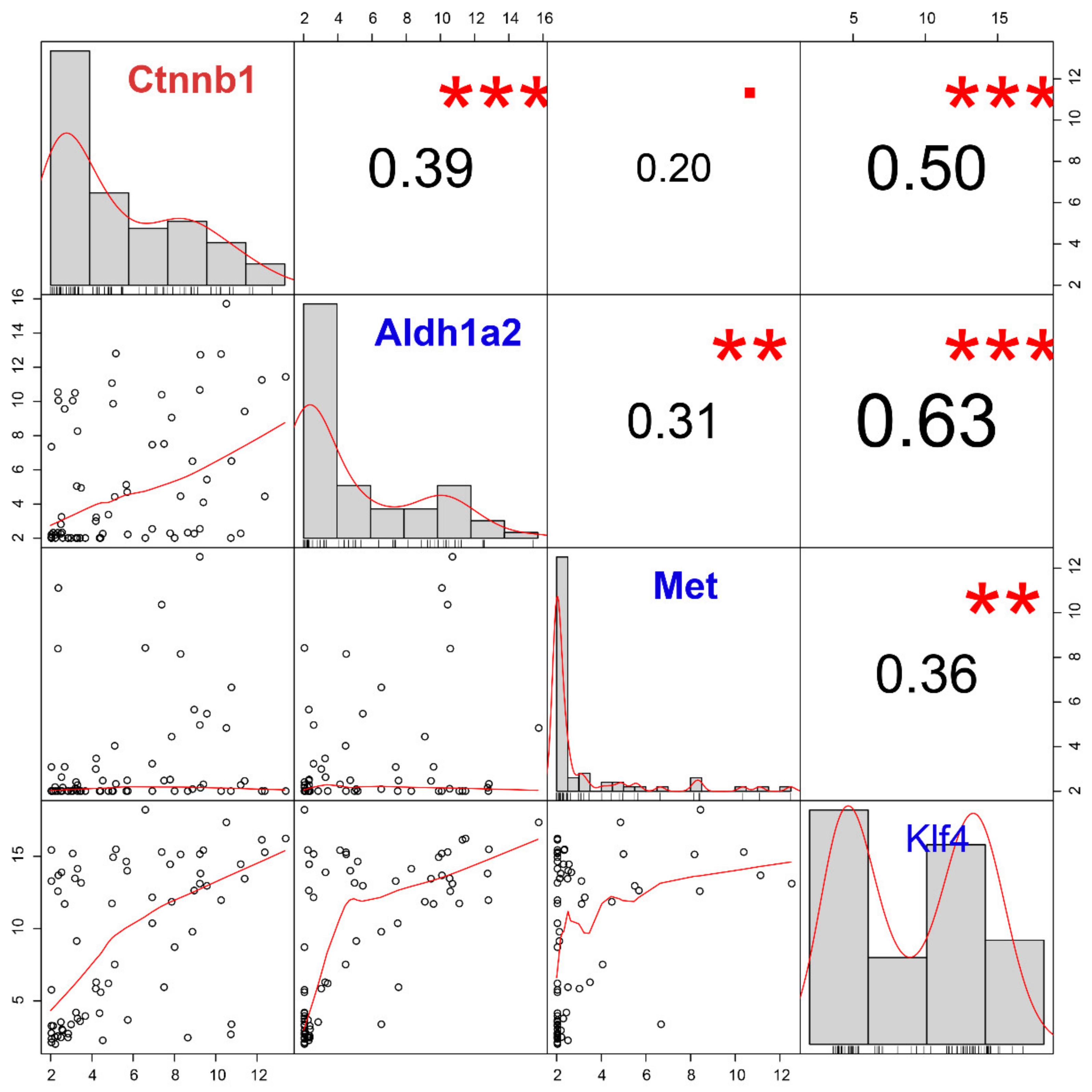
Wnt signaling is activated in pancreatic CTCs according to the original study of GSE51372 [
64]. Unexpectedly, we found no significant correlation between
Wnt family members and
Ctnnb1 in the pancreatic CTC-S group. In fact, according to our results, these
Wnt family members are even expressed at low levels (
Figure S6). Given that
Klf4 is overexpressed in the CTC-S group, we believe that
Klf4 inhibits
Wnt signaling in this context [
65]. Besides, the β-catenin has been determined to be necessary for cell adhesion and pluripotency, even without Wnt signaling [
66].
Klf4 (Krüppel-Like Factor 4) is enriched in CTC-S, which positively correlates with
Ctnnb1. As reported,
Klf4 is a versatile marker that can both suppress [
67] or support [
68] PDAC. Several studies have investigated
Klf4, which encodes a protein that belongs to the Krüppel family of transcription factors and plays a vital role in maintaining embryonic or induced pluripotent stem cells as well as in preventing their differentiation [
28,
69,
70]. In a recent study, Zheng et al. [
71] found that
Klf4 promotes CTC survival by increasing intracellular reactive oxygen species. Intriguingly,
Klf4 can interact with
Ctnnb1 through transcription regulation or direct protein interaction. As Tiwari et al. [
72] reported,
Ctnnb1 is one of the direct repression targets of the transcription factor
Klf4. On the opposite side, β-catenin could also bind with the promoter of
Klf4 through activating the canonical
Wnt pathway, as reported [
73]. However, we found that the canonical
Wnt family members are expressed at low levels, as previously mentioned. The evidence suggests that the interaction between
Klf4 and β-catenin might be happening through the formation of a protein complex rather than via the transcriptional regulation in the pancreatic CTC-S group. Even though there are studies claiming that
Klf4 inhibits β-catenin [
74,
75,
76], there are other reports showing that the
Klf4 / β-catenin complex is necessary for the self-renewal capacity of stem/cancer cells [
68]. The consensus is that the protein–protein interaction between
Klf4 and β-catenin happens in the nucleus [
68,
74,
75,
76]. According to previously published results [
76,
77], the cytoplasmic β-catenin could migrate to the nucleus freely, and there promote the transcription and translation of β-catenin targeted genes in conjunction with transcription factors. Among the β-catenin binding proteins extracted from epithelial cells nuclei, a 55-kDa protein could be identified as being
KLF4 [
76]. Furthermore, the de novo synthesis of both E-cadherin and β-catenin increased at a similar rate in order to reconstruct an adherens junction after the proteolytic disruption of this extracellular interaction in epithelial cancer cells [
77].
Clinically,
KLF4 expression in the primary tumor does significantly influence survival.
CTNNB1 is a negative prognostic marker for RFS and PFS. For OS, a trend can be seen, but it fails to reach statistical significance. This supports the hypothesis that the high expression of
Ctnnb1 in CTC-S contributes to the formation of adherens junctions, which in turn may promote the survival of CTC-S in circulation [
78]. However, it must be taken into account that the clinical validation was performed using RNA-seq expression data of bulk tumor cells from the primary tumor rather than single-cell RNA sequencing of CTCs, which limits its validity.
It is important to reinforce that this study is an in silico analysis of a single pre-existing dataset from a single pancreatic cancer model, since no other high-throughput sequencing datasets of pancreatic CTCs are publically available. To reduce the bias from limited study resources, we combined the analysis of differentially expressed genes, WGCNA, correlation analysis, and added clinical survival data. The limited sample size, and the absence of prognostic data directly associated with CTC expression data, may partially limit the results. With all that under consideration, more preclinical and clinical studies are necessary to confirm our findings. However, despite its exploratory nature, this study offers new insight into pancreatic CTCs with stem-like characteristics.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






