
Soluble Epoxide Hydrolase Inhibitor t-AUCB Ameliorates Vascular Endothelial Dysfunction by Influencing the NF-κB/miR-155-5p/eNOS/NO/IκB Cycle in Hypertensive Rats

Abstract
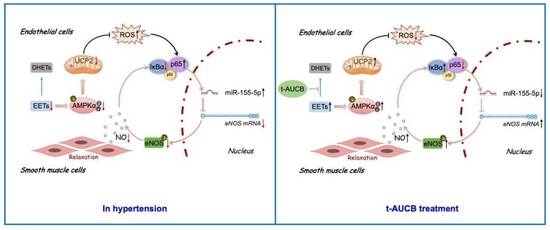
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and the 2K1C Model
2.2. Systolic Blood Pressure Measurement
2.3. Determination of Renal Blood Flow
2.4. Arterial Rings Preparation and Functional Assay
2.5. Primary Culture of Rat Renal Artery Endothelial Cells
2.6. Adenoviral Infection
2.7. Measurements of EETs
2.8. Treatment of Endothelial Cells
2.9. Western Blot Analysis
2.10. RNA Extraction and Quantitative Real-Time PCR
2.11. ROS Detection Using Dihydroethidium and MitoSOX Staining
2.12. Total Nitric Oxide (NO) Production
2.13. Statistical Analysis
3. Results
3.1. t-AUCB Restored Endothelial Function via Increasing EETs in 2K1C Hypertensive Rat Renal Arteries
3.2. t-AUCB Ameliorated Renal Arterial Endothelial Dysfunction by Stimulating AMPK in Hypertensive Rats
3.3. Uncoupling Protein 2 Mediated the Improvement of Endothelial Function, following AMPK Stimulation in Hypertensive Rats
3.4. Uncoupling Protein 2 Protected the Endothelial Function by Suppressing ROS/NF-κB in Hypertensive Rat Renal Arteries
3.5. MiR-155-5p Was Involved in the Improvement of Renal Arterial Endothelial Function by EETs in Hypertensive Rats
3.6. The AMPK/UCP2 Activation Regulated an NF-κB/miR-155-5p/eNOS/NO/IκB Cycle by Scavenging ROS in the Improvement of Endothelial Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olsen, M.H.; Angell, S.Y.; Asma, S.; Boutouyrie, P.; Burger, D.; Chirinos, J.A.; Damasceno, A.; Delles, C.; Gimenez-Roqueplo, A.P.; Hering, D.; et al. A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: The Lancet Commission on hypertension. Lancet 2016, 388, 2665–2712. [Google Scholar] [CrossRef]
- Dharmashankar, K.; Widlansky, M.E. Vascular endothelial function and hypertension: Insights and directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tan, N.; Zong, Y.; Li, L.; Zhang, Y.; Liu, J.; Wang, X.R.; Han, W.W.; Liu, L.L. LncRNA ENSMUST00000155383 is involved in the improvement of DPP-4 inhibitor MK-626 on vascular endothelial function by modulating Cacna1c-mediated Ca2+ influx in hypertensive mice. Front. Mol. Biosci. 2021, 8, 724225. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and endothelial function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Tang, F.; Lu, M.; Yang, J.; Han, R.; Mei, M.; Hu, J.; Wang, H. Pretreatment with Astragaloside IV protects human umbilical vein endothelialcells from hydrogen peroxide induced oxidative stress and cell dysfunction via inhibiting eNOS uncoupling and NADPH oxidase-ROS-NF-κB pathway. Can. J. Physiol. Pharm. 2016, 94, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef]
- Balduino Mendes, A.B.; Giollo-Junior, L.T.; de Andrade, D.O.; Gregório, M.L.; Yugar-Toledo, J.C.; Vilela-Martin, J.F. How to investigate the vascular changes in resistant hypertension. Curr. Hypertens. Rev. 2016, 12, 139–147. [Google Scholar] [CrossRef]
- Casanova, M.A.; Medeiros, F.; Trindade, M.; Cohen, C.; Oigman, W.; Neves, M.F. Omega-3 fatty acids supplementation improves endothelial function and arterial stiffness in hypertensive patients with hypertriglyceridemia and high cardiovascular risk. J. Am. Soc. Hypertens. 2017, 11, 10–19. [Google Scholar] [CrossRef]
- Di Daniele, N.; Marrone, G.; Di Lauro, M.; Di Daniele, F.; Palazzetti, D.; Guerriero, C.; Noce, A. Effects of caloric restriction diet on arterial hypertension and endothelial dysfunction. Nutrients 2021, 13, 274. [Google Scholar] [CrossRef]
- Ding, Y.; Tu, P.; Chen, Y.; Huang, Y.; Pan, X.; Chen, W. CYP2J2 and EETs protect against pulmonary arterial hypertension with lung ischemia-reperfusion injury in vivo and in vitro. Respir. Res. 2021, 22, 291. [Google Scholar] [CrossRef]
- Campbell, W.B.; Gebremedhin, D.; Pratt, P.F.; Harder, D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996, 78, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, J.; Wang, M.H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2016, 125, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Davis, B.B.; Jiang, D.Q.; Zhao, T.T.; Xu, D.Y. Soluble epoxide hydrolase inhibitors and cardiovascular diseases. Curr. Vasc. Pharmacol. 2013, 11, 105–111. [Google Scholar] [CrossRef]
- Duflot, T.; Roche, C.; Lamoureux, F.; Guerrot, D.; Bellien, J. Design and discovery of soluble epoxide hydrolase inhibitors for the treatment of cardiovascular diseases. Expert Opin. Drug Discov. 2014, 9, 229–243. [Google Scholar] [CrossRef]
- Verma, K.; Jain, S.; Paliwal, S.; Paliwal, S.; Sharma, S. A clinical perspective of soluble epoxide hydrolase inhibitors in metabolic and related cardiovascular diseases. Curr. Mol. Pharmacol. 2021, 15, 763–778. [Google Scholar] [CrossRef]
- Gao, J.; Bellien, J.; Gomez, E.; Henry, J.P.; Dautreaux, B.; Bounoure, F.; Skiba, M.; Thuillez, C.; Richard, V.J. Soluble epoxide hydrolase inhibition prevents coronary endothelial dysfunction in mice with renovascular hypertension. J. Hypertens. 2011, 29, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Davis, C.M.; Edin, M.L.; Lee, C.R.; Zeldin, D.C.; Alkayed, N.J. Role of endothelial soluble epoxide hydrolase in cerebrovascular function and ischemic injury. PLoS ONE 2013, 8, e61244. [Google Scholar] [CrossRef]
- Hanif, A.; Edin, M.L.; Zeldin, D.C.; Morisseau, C.; Falck, J.R.; Nayeem, M.A. Vascular endothelial over-expression of human soluble epoxide hydrolase (Tie2-sEH Tr) attenuates coronary reactive hyperemia in mice: Role of oxylipins and ω-Hydroxylases. PLoS ONE 2017, 12, e0169584. [Google Scholar]
- Chen, R.J.; Jiang, J.G.; **ao, X.; Wang, D.W. Effects of epoxyeicosatrienoic acids on levels of eNOS phosphorylation and relevant signaling transduction pathways involved. Sci. China C. Life Sci. 2005, 48, 495–505. [Google Scholar] [CrossRef]
- Jiang, J.G.; Chen, R.J.; **ao, B.; Yang, S.; Wang, J.N.; Wang, Y.; Cowart, L.A.; **ao, X.; Wang, D.W.; **a, Y.; et al. Regulation of endothelial nitric-oxide synthase activity through phosphorylation in response to epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 2006, 82, 162–174. [Google Scholar] [CrossRef]
- Nemecz, M.; Alexandru, N.; Tanko, G.; Georgescu, A. Role of microRNA in endothelial dysfunction and hypertension. Curr. Hypertens. Rep. 2016, 18, 87. [Google Scholar] [CrossRef]
- Zhang, H.N.; Xu, Q.Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, K.S.; Choi, S.; Kim, J.; Lee, D.K.; Park, M.; Park, W.; Kim, T.H.; Hwang, J.Y.; Won, M.H. NF-κB-responsive miRNA-31-5p elicits endothelial dysfunction associated with preeclampsia via down-regulation of endothelial nitric-oxide synthase. J. Biol. Chem. 2018, 293, 18989–19000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Yébenes, V.G.; Briones, A.M.; Martos-Folgado, I.; Mur, S.M.; Oller, J.; Bilal, F.; González-Amor, M.; Méndez-Barbero, N.; Silla-Castro, J.C.; Were, F.; et al. Aging-associated miR-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2408–2424. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, W.; Li, J.; Lu, J.; Lu, H.; Jia, W.; Liu, F. Exosomes derived from atorvastatin-pretreated MSC accelerate diabetic wound repair by enhancing angiogenesis via AKT/eNOS pathway. Stem Cell Res. Ther. 2020, 11, 350. [Google Scholar] [CrossRef]
- Gui, Y.; Chen, J.; Hu, J.; Liao, C.; Ouyang, M.; Deng, L.; Yang, J.; Xu, D. Soluble epoxide hydrolase inhibitors improve angiogenic function of endothelial progenitor cells via ERK/p38-mediated miR-126 upregulation in myocardial infarction mice after exercise. Exp. Cell Res. 2020, 397, 112360. [Google Scholar] [CrossRef]
- Liu, L.L.; Liu, J.; Wong, W.T.; Tian, X.Y.; Lau, C.W.; Wang, Y.X.; Xu, G.; Pu, Y.F.; Zhu, Z.M.; Xu, A.M.; et al. Dipeptidyl peptidase 4 inhibitor sitagliptin protects endothelial function in hypertension through a glucagon-like peptide 1-dependent mechanism. Hypertension 2012, 60, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.L.; Liu, J.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Xu, A.M.; Xu, G.; Ng, C.F.; Yao, X.Q.; Gao, Y.S.; et al. Uncoupling protein-2 mediates DPP-4 inhibitor-induced restoration of endothelial function in hypertension through reducing oxidative stress. Antioxid. Redox Signal. 2014, 21, 1571–1581. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, J.Y.; Park, M.; Kim, S.; Kim, T.; Kim, J.; Choi, S.; Park, W.; Hwang, J.Y.; Choe, J.; et al. NF-κB-dependent miR-31/155 biogenesis is essential for TNF-α-induced impairment of endothelial progenitor cell function. Exp. Mol. Med. 2020, 52, 1298–1309. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, J.Y.; Bae, J.; Kim, Y.M.; Won, M.H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Korean Red ginseng prevents endothelial senescence by downregulating the HO-1/NF-κB/miRNA-155-5p/eNOS pathway. J. Ginseng. Res. 2021, 45, 344–353. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial dysfunction and hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [PubMed]
- Higashi, Y.; Chayama, K. Renal endothelial dysfunction and hypertension. J. Diabetes Complicat. 2002, 16, 103–107. [Google Scholar] [CrossRef]
- Wang, D.; Borrego-Conde, L.J.; Falck, J.R.; Sharma, K.K.; Wilcox, C.S.; Umans, J.G. Contributions of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal afferent arterioles. Kidney Int. 2003, 63, 2187–2193. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Wu, M.Y.; Deng, B.Q.; Huang, J.; Hwang, S.H.; Li, M.Y.; Zhou, C.Y.; Zhang, Q.Y.; Yu, H.B.; Zhao, D.K.; et al. Inhibition of soluble epoxide hydrolase attenuates a high-fat diet-mediated renal injury by activating PAX2 and AMPK. Proc. Natl. Acad. Sci. USA 2019, 116, 5154–5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, D.; Tang, L.; Li, H.; Liu, Z.; Gao, J.; Edin, M.L.; Zhang, H.; Zhang, K.; Chen, J.; et al. Soluble epoxide hydrolase deficiency attenuates lipotoxic cardiomyopathy via upregulation of AMPK-mTORC mediated autophagy. J. Mol. Cell Cardiol. 2021, 154, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Contreras, C.; Sáenz-Medina, J.; Muñoz, M.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernandez, M.; López, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhang, H.; Wang, P.; Zhao, Y.; Li, Q.; Wei, X.; Cui, Y.; Sun, J.; Shang, Q.; Liu, D.; et al. Dietary curcumin ameliorates aging-related cerebrovascular dysfunction through the AMPK/uncoupling protein 2 pathway. Cell Physiol Biochem. 2013, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pu, Y.; Wang, P.; Chen, S.; Zhao, Y.; Liu, C.; Shang, Q.; Zhu, Z.; Liu, D. TRPV1-mediated UCP2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc Diabetol. 2013, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, X.; Huang, Q.; Li, S.; Zhou, Y.; Li, Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-κB pathway in vivo and in vitro. Redox Biol. 2018, 15, 62–73. [Google Scholar] [CrossRef]
- Pérez, L.; Vallejos, A.; Echeverria, C.; Varela, D.; Cabello-Verrugio, C.; Simon, F. OxHDL controls LOX-1 expression and plasma membrane localization through a mechanism dependent on NOX/ROS/NF-κB pathway on endothelial cells. Lab Invest. 2019, 99, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, C.; He, L.; Yang, D.; Cai, J.; Zhu, J.; Shu, S.; Liu, Y.; Yin, L.; Chen, G.; et al. The negative feedback loop of NF-κB/miR-376b/NFKBIZ in septic acute kidney injury. JCI Insight. 2020, 5, e142272. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Hamuro, M.; Sorescu, G.P.; Koyoma, K.; Sprague, E.A.; Jo, H.; Valente, A.J.; Prihoda, T.J.; Natarajan, M. IkappaBalpha-dependent regulation of low-shear flow-induced NF-kappa B activity: Role of nitric oxide. Am. J. Physiol Cell Physiol. 2003, 284, C1039–C1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Han, W.; Zhang, Y.; Zong, Y.; Tan, N.; Zhang, Y.; Li, L.; Liu, C.; Liu, L. Soluble Epoxide Hydrolase Inhibitor t-AUCB Ameliorates Vascular Endothelial Dysfunction by Influencing the NF-κB/miR-155-5p/eNOS/NO/IκB Cycle in Hypertensive Rats. Antioxidants 2022, 11, 1372. https://doi.org/10.3390/antiox11071372
Wang X, Han W, Zhang Y, Zong Y, Tan N, Zhang Y, Li L, Liu C, Liu L. Soluble Epoxide Hydrolase Inhibitor t-AUCB Ameliorates Vascular Endothelial Dysfunction by Influencing the NF-κB/miR-155-5p/eNOS/NO/IκB Cycle in Hypertensive Rats. Antioxidants. 2022; 11(7):1372. https://doi.org/10.3390/antiox11071372
Chicago/Turabian StyleWang, **aorui, Wenwen Han, Yi Zhang, Yi Zong, Na Tan, Yan Zhang, Li Li, Chang Liu, and Limei Liu. 2022. "Soluble Epoxide Hydrolase Inhibitor t-AUCB Ameliorates Vascular Endothelial Dysfunction by Influencing the NF-κB/miR-155-5p/eNOS/NO/IκB Cycle in Hypertensive Rats" Antioxidants 11, no. 7: 1372. https://doi.org/10.3390/antiox11071372