Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence

 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
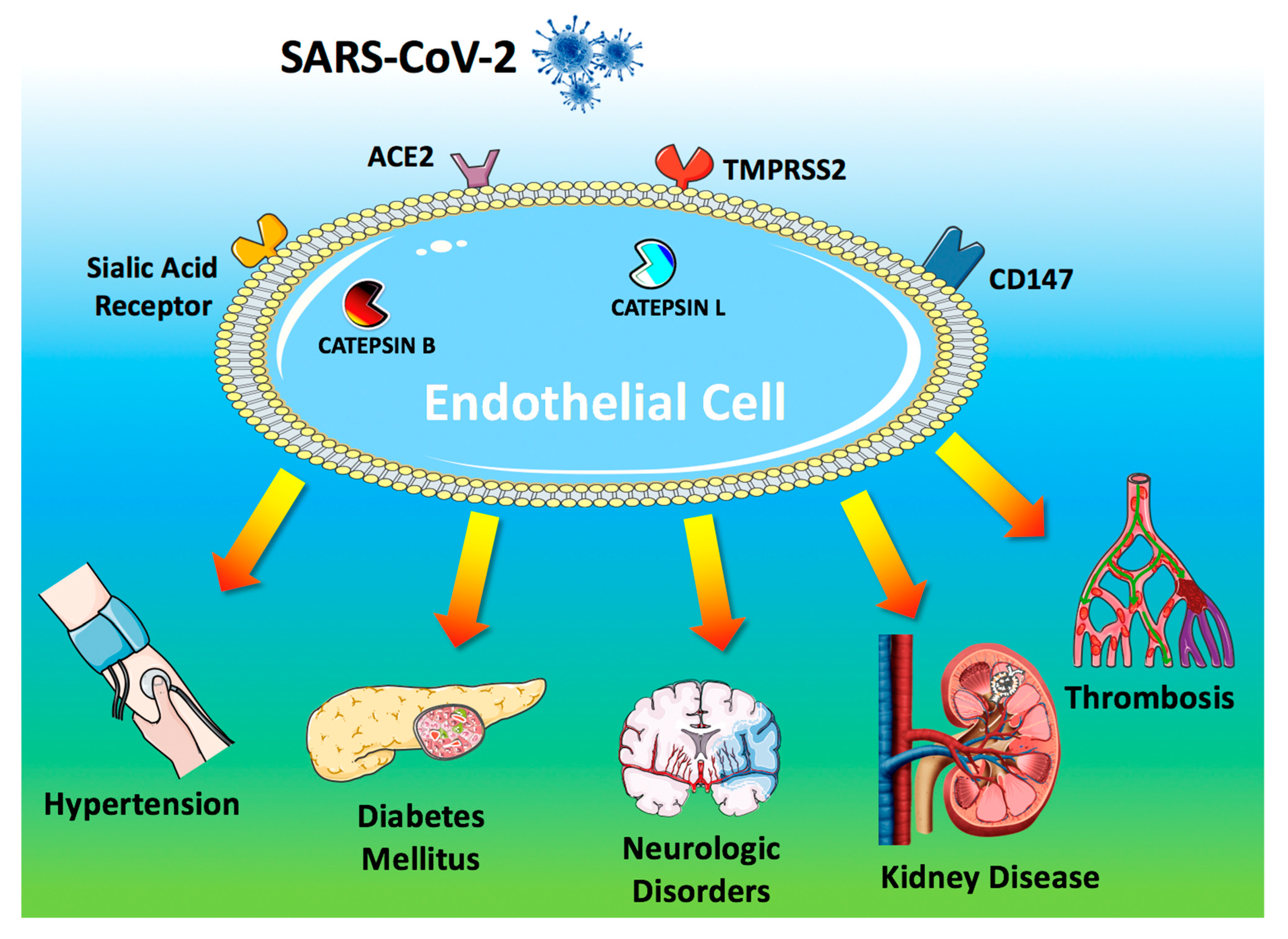
2. Pathogenesis of COVID-19
3. Hypertension and COVID-19
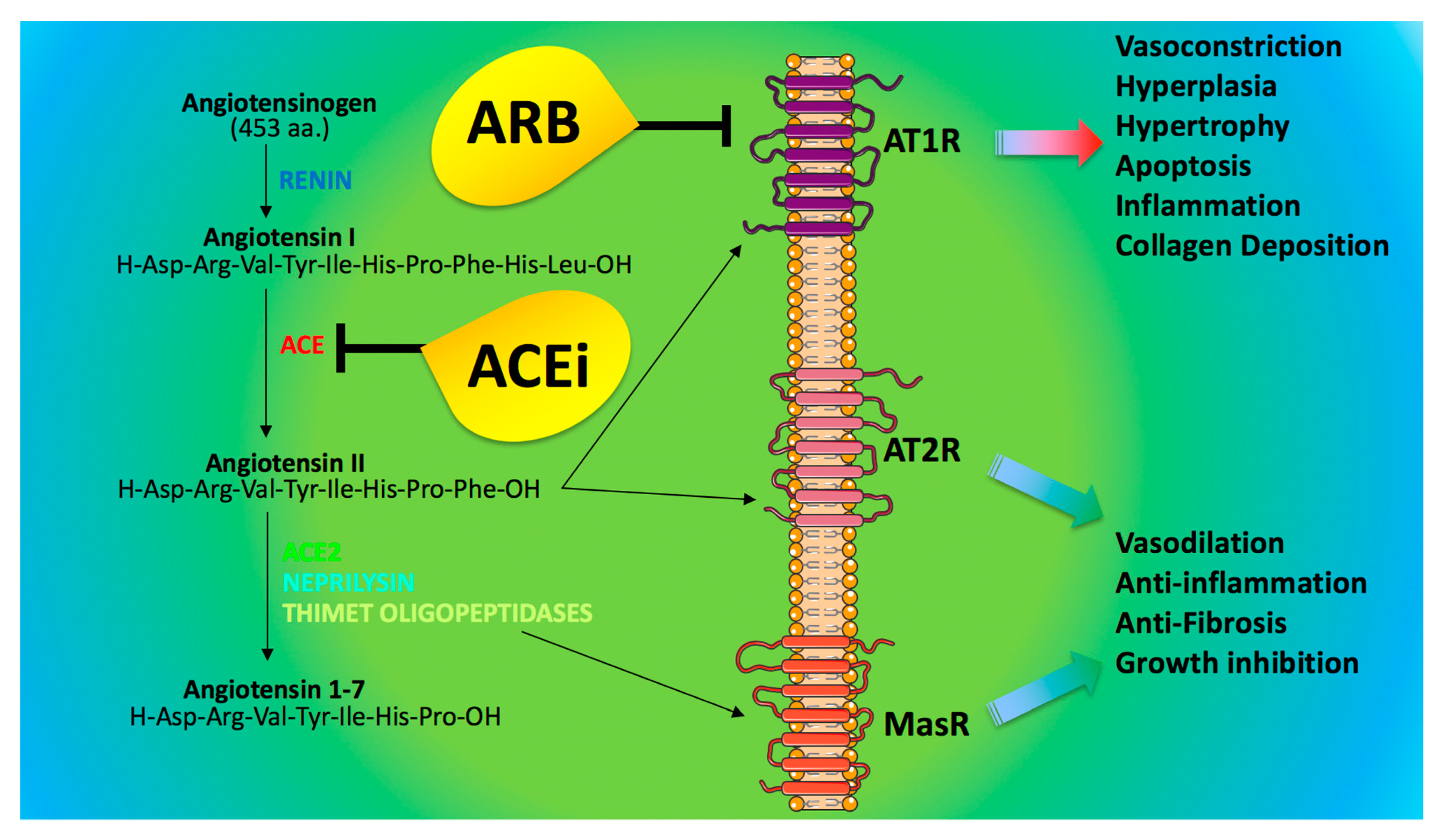
4. ACE2 and Anti-Hypertensive Drugs: What Do We Know?
5. Kidney Disease in COVID-19
6. Diabetes and COVID-19
7. Thromboembolism and COVID-19
8. Anticoagulation as a Key Therapy for COVID-19
Author Contributions
Funding
Conflicts of Interest
References
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More Than Just the Common Cold. JAMA 2020, 323, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, D.S.; Azhar, E.E.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Gasc, J.M.; Hamming, I.; van Goor, H.; Michaud, A.; van den Akker, L.H.; Jutten, B.; Cleutjens, J.; Bijnens, A.P.; Corvol, P.; et al. Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 2008, 215, 273–279. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Flack, J.; Ito, S.; Muntner, P.; Webb, C. Hypertension and COVID-19. Am. J. Hypertens. 2020, 33, 33–373. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W. The Northwell COVID-19 Research Consortium. Presenting Characteristics, Comorbidities, and Outcomes among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, L.C.; Parodi, S.M.; Escobar, G.J.; Liu, V.X. Characteristics of Hospitalized Adults With COVID-19 in an Integrated Health Care System in California. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with Covid-19 in China: A Nationwide Analysis. Eur. Respir. J. 2020, 2000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; ** Review and Meta-Analysis. Clin. Med. 2020, 9, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; De Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; ** review. Blood Rev. 2016, 30, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Sinha, P.; Delucchi, K.L.; McAuley, D.F.; O’Kane, C.M.; Matthay, M.A.; Calfee, C.S. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: A secondary analysis of randomised controlled trials. Lancet Respir. Med. 2020, 8, 247–257. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; **e, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Santulli, G.; Del Giudice, C.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Endothelial cells are able to synthesize and release catecholamines both in vitro and in vivo. Hypertension 2012, 60, 129–136. [Google Scholar] [CrossRef] [Green Version]
- **e, Y.; Wang, X.; Yang, P.; Shutong, Z. COVID-19 complicated by acute pulmonary embolism. Radiology 2020, 2, e200067. [Google Scholar] [CrossRef] [Green Version]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Heart J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Leonard-Lorant, I.; Delabranche, X.; Severac, F.; Helms, J.; Pauzet, C.; Collange, O.; Schneider, F.; Labani, A.; Bilbault, P.; Moliere, S.; et al. Acute Pulmonary Embolism in COVID-19 Patients on CT Angiography and Relationship to D-Dimer Levels. Radiology 2020, 201561. [Google Scholar] [CrossRef] [Green Version]
- Jolobe, O.M.P. Similarities Between Community-Acquired Pneumonia and Pulmonary Embolism. Am. J. Med. 2019, 132, e863. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Yang, J.; Santulli, G.; Reiken, S.R.; Wronska, A.; Kim, M.M.; Osborne, B.W.; Lacampagne, A.; Yin, Y.; Marks, A.R. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc. Natl. Acad. Sci. USA 2016, 113, 8532–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gando, S.; Levi, M.; Toh, C.H. Disseminated intravascular coagulation. Nat. Rev. Dis. Primers. 2016, 2, 16037. [Google Scholar] [CrossRef] [PubMed]
- Walborn, A.; Rondina, M.; Mosier, M.; Fareed, J.; Hoppensteadt, D. Endothelial Dysfunction Is Associated with Mortality and Severity of Coagulopathy in Patients with Sepsis and Disseminated Intravascular Coagulation. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619852163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.J.; Baden, L.R.; Morrissey, S. Audio Interview: New Research on Possible Treatments for Covid-19. New Engl. J. Med. 2020, 382, e30. [Google Scholar] [CrossRef]
- Ahn, D.G.; Shin, H.J.; Kim, M.H.; Lee, S.; Kim, H.S.; Myoung, J.; Kim, B.T.; Kim, S.J. Current Status of Epidemiology, Diagnosis, Therapeutics, and Vaccines for Novel Coronavirus Disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 202005615. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Limon, P.; Ortega, R.; Arias de la Rosa, I.; Abalos-Aguilera, M.D.C.; Perez-Sanchez, C.; Jimenez-Gomez, Y.; Peralbo-Santaella, E.; Font, P.; Ruiz-Vilches, D.; Ferrin, G.; et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl. Res. 2017, 183, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Higashi, Y.; Tomiyama, H.; Maruhashi, T.; Kurisu, S.; Kihara, Y.; Mutoh, A.; Ueda, S.I. Effect of short-term colchicine treatment on endothelial function in patients with coronary artery disease. Int. J. Cardiol. 2019, 281, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Parchure, N.; Zouridakis, E.G.; Kaski, J.C. Effect of azithromycin treatment on endothelial function in patients with coronary artery disease and evidence of Chlamydia pneumoniae infection. Circulation 2002, 105, 1298–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Chen, B.; Zhao, Z.; He, N.; Zeng, Z.; Wu, B.; Fukushima, Y.; Dai, M.; Huang, Q.; Xu, D.; et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res. Cardiol. 2013, 108, 342. [Google Scholar] [CrossRef] [PubMed]
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Le, N.T.; Takei, Y.; Izawa-Ishizawa, Y.; Heo, K.S.; Lee, H.; Smrcka, A.V.; Miller, B.L.; Ko, K.A.; Ture, S.; Morrell, C.; et al. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J. Immunol. 2014, 193, 3803–3815. [Google Scholar] [CrossRef]
- Rahman, R.; Murthi, P.; Singh, H.; Gurusinghe, S.; Mockler, J.C.; Lim, R.; Wallace, E.M. The effects of hydroxychloroquine on endothelial dysfunction. Pregnancy Hypertens. 2016, 6, 259–262. [Google Scholar] [CrossRef]
- Gambardella, J.; Morelli, M.; Sardu, C.; Santulli, G. Targeting Endothelial Dysfunction in COVID-19. Nature 2020, in press. [Google Scholar]
- Ciccarelli, M.; Santulli, G.; Campanile, A.; Galasso, G.; Cervero, P.; Altobelli, G.G.; Cimini, V.; Pastore, L.; Piscione, F.; Trimarco, B.; et al. Endothelial alpha1-adrenoceptors regulate neo-angiogenesis. Br. J. Pharmacol. 2008, 153, 936–946. [Google Scholar] [CrossRef]
- Wilbert-Lampen, U.; Seliger, C.; Zilker, T.; Arendt, R.M. Cocaine increases the endothelial release of immunoreactive endothelin and its concentrations in human plasma and urine: Reversal by coincubation with sigma-receptor antagonists. Circulation 1998, 98, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.S.; McKeown, L.; Tumova, S.; Liu, R.; Seymour, V.A.; Wilson, L.A.; Naylor, J.; Greenhalgh, K.; Hou, B.; Majeed, Y.; et al. Inhibition of endothelial cell Ca(2)(+) entry and transient receptor potential channels by Sigma-1 receptor ligands. Br. J. Pharmacol. 2013, 168, 1445–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massamiri, T.; Duckles, S.P. Sigma receptor ligands inhibit rat tail artery contractile responses by multiple mechanisms. J. Pharmacol. Exp. Ther. 1991, 259, 22–29. [Google Scholar] [PubMed]
- Hamidi Shishavan, M.; Henning, R.H.; van Buiten, A.; Goris, M.; Deelman, L.E.; Buikema, H. Metformin Improves Endothelial Function and Reduces Blood Pressure in Diabetic Spontaneously Hypertensive Rats Independent from Glycemia Control: Comparison to Vildagliptin. Sci. Rep. 2017, 7, 10975. [Google Scholar] [CrossRef] [Green Version]
- Bolz, S.S.; Pohl, U. Indomethacin enhances endothelial NO release--evidence for a role of PGI2 in the autocrine control of calcium-dependent autacoid production. Cardiovasc. Res. 1997, 36, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Sfikakis, P.P.; Papamichael, C.; Stamatelopoulos, K.S.; Tousoulis, D.; Fragiadaki, K.G.; Katsichti, P.; Stefanadis, C.; Mavrikakis, M. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum. 2007, 56, 1985–1993. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation with In-Hospital Survival Among Hospitalized Patients with COVID-19. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Basu, A.; Kanda, T.; Beyene, A.; Saito, K.; Meyer, K.; Ray, R. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 2007, 81, 3933–3941. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Pavy, M.; Young, N.; Freeman, C.; Lobigs, M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral. Res. 2006, 69, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.J.; Pizzato, M.; Takeuchi, Y.; Devereux, S. Heparin binds to murine leukemia virus and inhibits Env-independent attachment and infection. J. Virol. 2002, 76, 6909–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, B.J.; Lortat-Jacob, H. Human immunodeficiency virus and heparan sulfate: From attachment to entry inhibition. Front. Immunol. 2013, 4, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahmias, A.J.; Kibrick, S. Inhibitory effect of heparin on herpes simplex virus. J. Bacteriol. 1964, 87, 1060–1066. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. https://doi.org/10.3390/jcm9051417
Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. Journal of Clinical Medicine. 2020; 9(5):1417. https://doi.org/10.3390/jcm9051417
Chicago/Turabian StyleSardu, Celestino, Jessica Gambardella, Marco Bruno Morelli, Xujun Wang, Raffaele Marfella, and Gaetano Santulli. 2020. "Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence" Journal of Clinical Medicine 9, no. 5: 1417. https://doi.org/10.3390/jcm9051417