1. Introduction
Parasitosis, which is infection with parasites, is a prevalent cause of morbidity among humans worldwide [
1,
2,
3]. Tropical zones, particularly those that are impoverished, conflicted, or unsanitary, serve as endemic foci for a range of parasitic diseases [
3,
4,
5]. The World Health Organization (WHO) has reported that annually 48.4 million cases and 59,724 deaths are attributed to the prevalence of 14 parasites, accounting for a total burden of 8.78 million disability-adjusted life years (DALYs). Of these, 48% represent foodborne parasitic diseases, accounting for 76% of the DALYs [
6]. Transmission through contaminated food is prevalent in low- and middle-income countries [
6]. Approximately 241 million cases of malaria and 627,000 deaths resulting from malaria were reported globally in 2020. Innumerable deaths are caused by other parasitic infections, most notably neglected tropical diseases (NTDs) [
2,
5,
7].
Unlike the vast majority of bacterial and viral infections, which have an incubation period ranging from a few hours to days, parasitic diseases tend to have an incubation period of weeks or even months. The incubation period of specific parasitic diseases, like alveolar echinococcosis, can extend up to 10 years [
2,
8]. Therefore, early and precise detection of parasitosis is imperative for timely curative interventions and prevention of pandemics. However, current reliable or commonly used detection methods are limited by sensitivity, reaction time, and equipment dependence to achieve this purpose [
9,
10].
A promising new method for nucleic acid detection utilizes the CRISPR-associated (Cas) nuclease, which can overcome the limitations of instrument dependence and laborious operational processes [
11,
12,
13,
14,
15]. By meticulously selecting target genes and designing specific CRISPR crRNAs (crRNAs), precise detection of various parasites can be guaranteed [
13,
15,
16,
17]. Due to the precise recognition capability of crRNA, a broad operational temperature range, and an intuitive result observation method, it is progressively evolving into an optimal tool for on-site testing [
18,
19,
20,
21]. When used with thermostatic amplification techniques like RPA and LAMP, CRISPR/Cas12a shows higher specificity and sensitivity [
22,
23,
24,
25,
26,
27]. The main objective of testing in field environments is to decrease the number of devices and simplify transportation conditions while maintaining both specificity and sensitivity. To improve the applicability and effectiveness of parasite monitoring in the field, continuous optimization of existing one-pot detection systems and the development of convenient biosensors that combine all essential steps into one are crucial.
The present review compares the CRISPR/Cas12a system with alternative molecular methods for the detection of parasitic diseases. Emphasis is on enhancement of the one-pot recombinase polymerase amplification (RPA)-CRISPR/Cas12a and improvement of CRISPR/Cas12b or Cas13 assays.
2. Application of Nucleic Acid Amplification Tests in Parasite Detection
Currently, the diagnosis of parasitic diseases relies on several approaches, such as epidemiology and pathophysiology, and methods including microscopy, immunodiagnostics, and nucleic acid amplification tests (NAATs). Among these, the microscopic detection of parasites remains the most reliable [
9,
28]. However, in underdeveloped regions with high rates of parasitosis, skilled microscope operators are often scarce, making this technique challenging to implement [
29]. Furthermore, this approach is unsuitable for conditions linked to parasites at different developmental stages, which pose challenges in their detection within blood or stool specimens.
Immunoassay-based diagnostic procedures have been used for decades and are widely used for detecting parasites. However, their application for diagnosis of parasitosis has been limited due to several drawbacks including the possibility of false negatives and false positives [
30,
31,
32].
Molecular detection of nucleic acids demonstrates superior sensitivity, specificity, and reproducibility compared to alternative methods (
Table 1). Consequently, NAATs are preferred molecular detection tools due to their ability to amplify trace amounts of DNA and RNA, allowing for highly specific detection by complementary nucleotide pairing [
2,
9]. Polymerase chain reaction (PCR) is currently the most prevalent NAAT tool. Among them, quantitative real-time PCR (qPCR) has demonstrated the best sensitivity and specificity in the detection of various parasites and digital PCR (dPCR) can be powerful in quantifying nucleic acids [
2,
33,
34,
35,
36]. Furthermore, a PCR-ELISA-based detection technique has been established, reducing the limit of detection (LOD) to 0.3 fg, equivalent to 0.004 parasites; however, this method takes longer than 4 h [
37]. Although these techniques have been instrumental in establishing dependable diagnostic methods for parasitosis, including malaria, filariasis, toxoplasmosis, and echinococcosis, they require prolonged reaction times, intricate handling, expensive laboratory equipment, and a high level of technical expertise [
10,
38,
39,
40,
41].
Isothermal amplification has been employed in the diagnosis of various parasitic diseases, addressing the challenges posed by traditional diagnostic methods [
42,
43,
44,
45,
46]. Compared to PCR, isothermal amplification technology, exemplified by loop-mediated isothermal amplification (LAMP) and RPA, significantly reduces the reaction time and dependence on instruments. RPA is an efficient method for on-site detection due to its simple primer design, low-temperature requirements, and easy storage [
43,
44]. Recombinase-aid amplification (RAA), based on the same principle, also offers these advantages in rapid detection [
47]. The assay results presentation has transitioned from gel electrophoresis to using fluorescence, turbidity, color, and lateral flow, which are easier to manipulate and observe, thus enhancing their field operation applicability [
42,
48,
49].
In various laboratories and regions, qPCR remains the primary or sole standard in NAAT due to issues with standardizing other assays such as PCR, RPA, and LAMP [
50]. Determining the reliability of results and setting a reliable assay time are among the challenges. Sequencing is a common method of validation, but it significantly extends the time required to obtain assay results. Environmental factors, such as temperature and humidity, may also limit the application of these technologies for field monitoring by affecting the stability and reliability of the reagents. To address this issue, sealed lyophilized powders can be used to preserve the reagents [
20].
In addition to the selection of appropriate detection methods for NAATs, which has a significant impact on the accuracy and sensitivity of diagnosis of parasitosis, the selection of target genes is a key consideration. Along with 18S ribosomal RNA (rRNA), Internal Transcribed Spacer (ITS), and mitochondrial genes, stable tandem repeats have recently come into focus. In most parasite genomes, repetitive sequences make up a substantially greater proportion compared to coding sequences, comprising an estimated 20% or even surpassing 30% [
51,
52]. Numerous tandem repeats have been used to detect multiple protozoans and worms, such as
Trypanosoma cruzi,
Onchocerca volvulus, and
Schistosoma mansoni (
Table 2).
Point-of-care testing (POCT), a priority for strategies relying on mass drug administration to control several NTDs, is a medical diagnostic tool that can be used near or at the point-of-care, allowing for on-site testing [
20,
76]. In ideal POCT, the steps required to go from raw sample to understandable result should be minimized, enabling unskilled operators to perform the analysis. Therefore, it is imperative to develop nucleic acid-based diagnostic tools that combine the sensitivity and specificity of established NAATs with the convenience, cost-effectiveness, and speed of isothermal amplification-based POCT methods. CRISPR-based diagnostics have the potential to fulfill all these requirements (
Table 3).
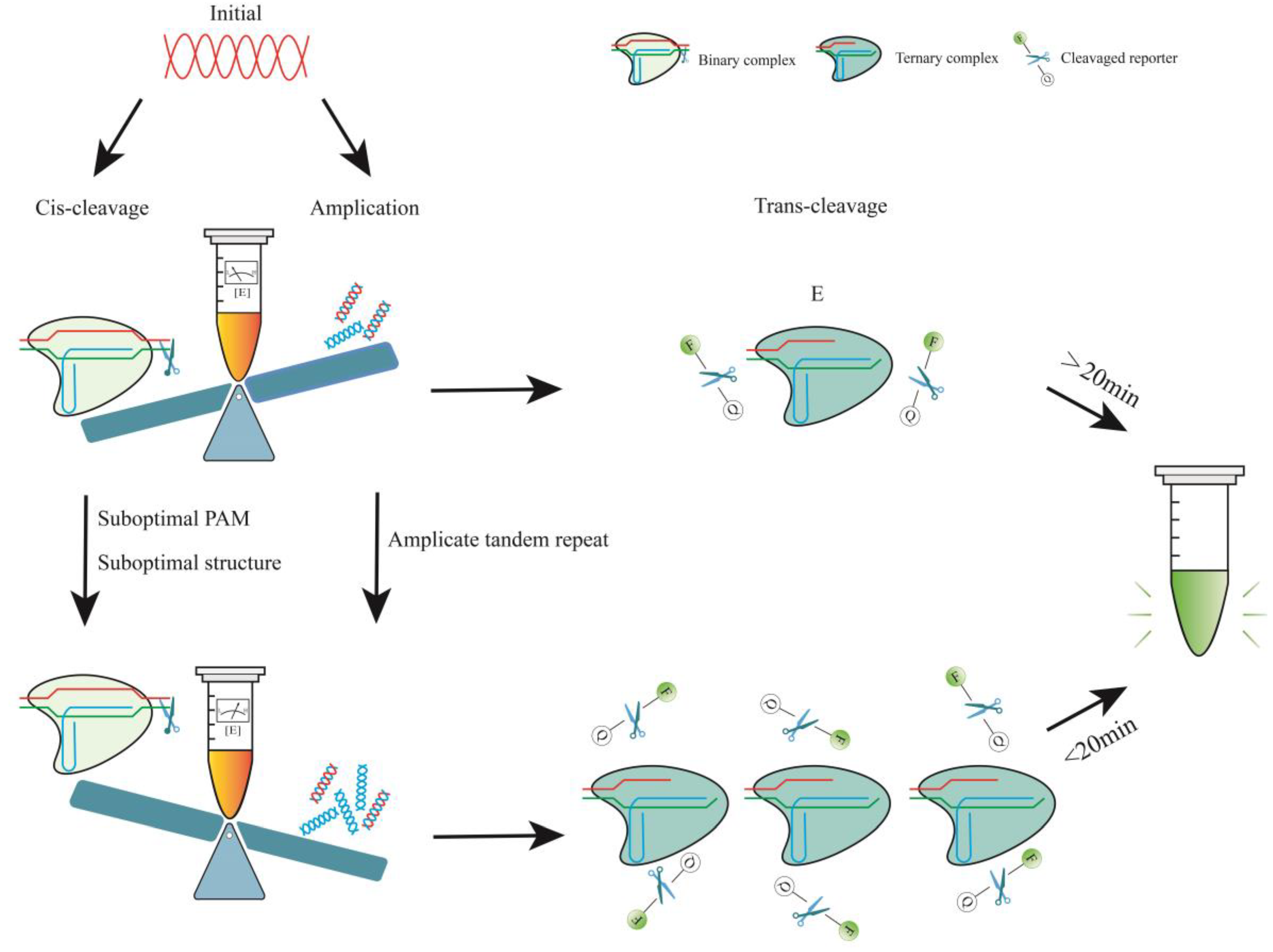
4. Optimization of the CRISPR/Cas12a One-Pot Detection Assay
Due to the low initial concentration of the target gene in a sample and kinetic rates that result in an amplification-free LOD in the picomolar range under standard assay conditions [
150], Cas12a detection often requires an amplification process before application. This implies that the signal amplification is usually conducted in two processes [
109]. Target genes are initially amplified using RPA or LAMP techniques. The resulting amplified products are subsequently transferred to the Cas12a system for cleavage, followed by fluorescence signal generation.
To streamline operations and prevent cross-contamination during field tests, the one-pot method is now predominantly utilized. This assay allows the simultaneous amplification and cleavage of Cas12a. Freeze-drying all components and integrating them into the sensor ensures consistent performance in various environmental conditions [
27,
125]. However, this leads to the cis-cleavage of Cas12a, which reduces the concentration of the target genes while RPA enhances it. Therefore, it is crucial to optimize amplification in the initial phase of the reaction.
POCT sensors that use amplification followed by detection strategy often require multiple liquid transfers during testing, which reduces their user-friendliness [
20]. The CRISPR/Cas12a one-pot detection system shows promise in replacing reagents in more mature amplification-free POC sensors, which can further improve the sensitivity of detection. However, significant challenges remain in terms of cost and complexity of devices.
4.1. One-Pot One-Step Reaction
4.1.1. Determinants of Cas12a Enzyme Kinetics
Several studies have reported rapid single-turnaround, cis-cleavage reactions at low target concentrations, with typical reaction times of approximately 100 s [
151]. A Michaelis–Menten model for Cas12a trans-cleavage activity was established and validated by a team from Stanford University. This was achieved through the utilization of varying concentrations of substrates, targets, and crRNAs [
150,
151]. The authors suggest that the concentration of the trans-cleavage product formed over time can be described using the following scaling equation:
The production efficiency of the trans-cleavage product P is influenced by both reaction time and τ. To refer to the target-activated Cas12-crRNA-target DNA complex, we use E, and subsequently, [E] represents the concentration of this complex. The characteristic time required to complete trans-cleavage is governed by the time scale τ, which is proportional to
KM and inversely proportional to
kcat and [E] [
151]. The rate constant
kcat/
KM of enzymatic reactions is affected by the Cas type, crRNA, incubation time, pH, and temperature [
150]. During the early stages of the reaction, [E] equals the concentration of the target molecule (c), which depends on c
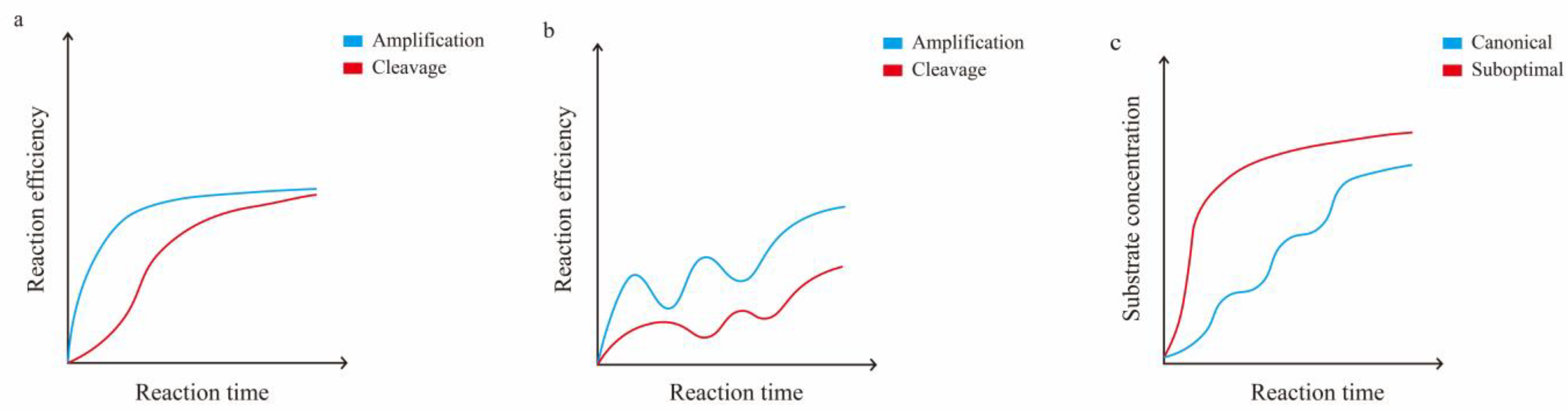
0, amplification, and cis-cleavage. Therefore, one could use suboptimal crRNA to weaken cis-cleavage or employ other methods to ensure that amplification dominates the pre-reaction period, resulting in a rapid increase of [E] (
Figure 2).
In the CRISPR/Cas12a system, the crRNA binds to the Cas12a effector protein to form a binary complex (ribonucleoprotein). This complex then locates the PAM sequence and verifies the adjacent spacer sequence, thereby initiating both cis- and trans-cleavage [
91,
94]. When using crRNAs with suboptimal structures or suboptimal PAMs, their cleavage activity may be impaired by affecting the efficiency of binding or recognition [
22]. With these methods, amplicons can be rapidly accumulated for the activation of large numbers of Cas12a–crRNA–target ternary complexes (
Figure 2). Sensitivity and detection time were significantly improved without compromising specificity. It is important to note that suboptimal crRNAs will also affect their collateral activity, thereby reducing the rate of fluorescence signal growth. Therefore, it is necessary to fully compare and screen the use of such suboptimal crRNAs.
4.1.2. Reduced crRNA Efficiency by PAM
In the CRISPR-Cas system, the effector nuclease must identify the PAM adjacent to the target site for initiation of target recognition [
152]. Studies of the crystal structure of the LbCas12a–crRNA binary complex [
153] and AsCas12a–crRNA–target DNA ternary complex [
94,
154] reveal the mechanisms involved in Cas12a and crRNA recognition, as well as the operations of crRNA-directed DNA targeting and PAM recognition. In Cas12a, the PAM duplex is enveloped within a PAM-binding channel formed by the WED, REC1, and PI domains. The sequence and conformation of PAM duplexes are primarily recognized by two conserved lysine residues (i.e., base and shape readout mechanism) [
155]. These findings suggest that the PAM-binding channel of Cas12a is flexible in conformation, allowing for the identification of both canonical and non-canonical PAMs [
155]. LbCas12a and AsCas12a identify TTTV and CTTV/TCTV/TTCV as canonical and suboptimal PAM, respectively [
155,
156].
In 2022, a Chinese team conducted a one-pot test called sPAMC, which refers to a suboptimal PAM of a Cas12a-based test [
22]. A comparison of the collateral activity revealed that crRNAs utilizing suboptimal PAMs demonstrated lower potency and slower kinetics in comparison to those utilizing canonical PAMs. Nevertheless, over 80% of the 120 suboptimal PAMs displayed quicker reactions than canonical PAMs in the one-pot reactions.
The appearance of the target amplicon was observed 2 min after the one-pot reaction utilizing suboptimal PAM, in contrast to the 8–10 min required for canonical PAM. Utilizing a suboptimal PAM with varied concentrations of Cas12a/crRNA ribonucleoprotein yielded steady kinetic curves, in contrast to reactions with traditional PAMs. In one-pot reactions, several uncommon PAMs (such as VTTV, TCTV, and TTVV) and some TRTV, TTNT, and YYYN PAMs (excluding TTTV) outperformed canonical PAMs. The SARS-CoV-2 diagnostic method established using suboptimal PAM demonstrates a sensitivity comparable to that of qPCR, with a reaction time of merely 15 min [
22]. However, it seems that this approach is not applicable to AapCas12b [
157]. This may be due to the PAM sequence of AapCas12b (TTN) being potentially less adaptable than that of LbCas12a (TTTV), and even a single nucleotide mutation within AapCas12b’s PAM could significantly impair its activity [
157].
Substituting residues within the PAM-interacting domain of Cas enzyme can achieve a similar effect in adjusting its activity. This idea has been applied to Cas12b by the same team and proved to be effective [
157].
4.1.3. Reduced crRNA Efficiency by Structure
Suboptimal crRNAs can be selected based on their structure while ensuring specificity. If CRISPR/Cas9 cleavage is an energy-driven process, its efficiency relies substantially on nucleotide hybridization and changes in folding-free energy [
158,
159]. The stability of guide RNA (gRNA)-DNA for gRNAs exhibiting different efficiencies significantly varies. When local sliding is examined, an energy model accurately predicts the efficiency of gRNAs. In CRISPR/Cas12a, research has shown that the activity of the Cas12a system is positively correlated with the stable binding between the activator and the crRNA [
160]. The structure of the single ssDNA activator has also been found to affect the Cas12a trans-cleavage activity [
160,
161]. Furthermore, engineering a hairpin secondary structure in the crRNA spacer region can greatly improve its specificity [
162]. Therefore, it is crucial to consider the use of suboptimal crRNAs when develo** a one-pot detection method. Additionally, the efficiency of the RPA-CRISPR/Cas12a one-pot and one-step reaction can also be enhanced by using crRNAs that are not restricted by the PAM sequence [
117] or by reducing the dosage of Cas12a [
163].
4.2. One-Pot Reactions with Two Steps
4.2.1. Light-Activated crRNA to Initiate Cleavage
Control of chemical reactions through photocontrolled techniques can be achieved in a non-contact manner within seconds. This technology has been used extensively in both CRISPR/Cas9 research and practice [
164,
165,
166], and has also been progressively refined for CRISPR/Cas12a detection [
167]. Initially, the CRISPR/Cas12a system is blocked by a photo-cleaved linker containing crRNA to ensure optimal RPA performance. After amplification, the Cas12a detection system is activated via light to initiate trans-cleavage and produce fluorescence signals [
167]. However, the constant optimization of the ratio between the photocleaved linker and crRNA, along with the compromised stability of the Cas12a-crRNA complex due to the lack of pre-binding of crRNA to Cas proteins, hinders the effectiveness of this approach.
The same group of researchers subsequently developed a novel CRISPR/Cas12a detection assay that uses 6-nitropiperonyloxymethyl-caged thymidine (NPOM-dt) to modify crRNA [
168]. This method involves caging crRNA to prevent base pairing between itself and the target, rather than binding it to the Cas enzyme. Rapid activation can be attained by photoinduced decaying, which makes this approach simpler, faster, and more stable. It should be noted that optimization of the irradiation time and the number and position of NPOM may need reconsideration for different pathogens. In the context of POCT, challenges persist with reagent storage conditions, actual amplification time, and the portability of illumination devices.
4.2.2. Physical Separation of the Two Processes
In addition to performing two reactions simultaneously in one tube, it is also possible to physically separate the two reaction systems in one tube, to allow for sequential progression [
20,
169,
170,
171]. The CRISPR/Cas12a reagents are spun down for cleavage after DNA amplification by leveraging the enhanced surface tension of the protein-containing liquid [
172]. Initially, the RPA reaction takes place at the base of the tube, while CRISPR/Cas12a is positioned at the lid, separate from the reaction. After amplification for 20 min, the CRISPR/Cas12a reagent is briefly spun into the reaction mixture without opening the tube. The reaction continues, and the RPA amplicon activates the Cas12a nuclease to trans-cleave the fluorescent ssDNA-FQ molecule, resulting in a fluorescent signal. However, this method is cumbersome, particularly in the context of large-scale POCT.
In brief, cis-cleavage plays a crucial role as the rate-determining step for overall performance in one-pot reactions [
22,
151]. During the initial stage, low-concentration targets are diminished due to cis-cleavage, which results in a slow and unstable accumulation of amplicons. Consequently, the growth of the signal decreases or may even disappear altogether (
Figure 3). The
kcat/
KM of the enzyme can be reduced by utilizing a suboptimal PAM or structure, which slows cis-cleavage. This results in a balance between the two-signal amplification processes of RPA and trans-cleavage. Through careful engineering of enzyme engineering [
157,
173], primer design, crRNA design [
22,
121,
174,
175], reaction system [
125,
130,
174,
176,
177,
178,
179,
180], and reporter selection [
181,
182], isothermal amplification and CRISPR detection can be effectively combined in a one-pot reaction [
50]. The optimized Cas12a assay even has the potential to achieve the same detection performance at room temperature [
157].
5. Conclusions
Parasites are prevalent in the natural world, particularly in underdeveloped regions, and result in high DALYs and substantial economic losses. This necessitates the development of rapid, sensitive, and accurate diagnostic tools for detecting parasites. The emergence of CRISPR, and specifically recent examinations of Cas12a, compensates for the limitations of isothermal amplification and presents a fresh approach for POCT. With the collateral activity of Cas12a, results can be evaluated intuitively via the inclusion of fluorophores. Combined with RPA, samples with even small numbers of pathogens can be quickly and accurately tested at the POC.
For POCT, the one-pot method is the best option due to its ability to prevent cross-contamination and the significant simplicity of the procedure. Nonetheless, current one-pot detection techniques are associated with several limitations, including extended reaction times, low sensitivity, complicated operation, and reliance on sample pretreatment. Additionally, the utilization of RPA has restricted the advancement of CRISPR assays to some extent. As the most commonly used partner for the CRISPR one-pot method, RPA kits are only sold by a few companies, with high prices and an unstable supply [
48].
By balancing the two processes of amplification and cleavage with a suboptimal PAM or structure, the detection performance of the one-pot method can be improved. With suboptimal conditions, the limitation of PAM or crRNA on target genes can also be reduced, thereby expanding the pool of target genes. In addition, light-activated crRNA and spatial isolation enable two reactions to proceed in one-pot sequentially without the requirement for opening the lid. Furthermore, incorporating tandem repeats as targets can significantly enhance the amplification efficiency and sensitivity of detection, regardless of sample preparation methods. These modifications can potentially enhance not only RPA-Cas12a but also all Cas12a detection methods involving amplification. Furthermore, it is important to assess these concepts not only concerning Cas12a, but also in other CRISPR systems, including Cas12b, Cas13, and even Cas9.
The samples used for parasite testing are primarily blood and feces, and their nucleic acid extraction often relies on silica gel column chromatography that takes at least 45 min. In POCT, there is an increasing need for nucleic acid extraction methods that provide shorter operating times, simpler devices, and products with minimal inhibitors. Several POC nucleic acid extraction platforms have been developed to meet these requirements, including microfluidic chips, paper-based devices, microneedle patches, digital microfluidics, and hand-operated microfluidic systems [
183]. However, these techniques are primarily focused on bacteria and viruses and have not been validated for parasite detection, and present challenges in extraction of high-quality RNA.
Although the sensitivity and accuracy of most CRISPR-based assays greatly exceed those of antigen test kits and isothermal amplification assays, which are currently more suitable for POCT, CRISPR still cannot replace them as the preferred choice for POCT, either due to their high cost or due to the complex storage and transportation conditions. In the future, for the application of CRISPR/Cas12a to POCT, it is necessary to continually optimize the one-pot method detection efficiency and identify a more compatible isothermal amplification technology. Alternatively, the existing amplification-free CRISPR/Cas detection technology can be further optimized [
121,
177,
179,
184].
Future studies should include larger-scale CRISPR/Cas12a clinical assay experiments and validation of sensor stability to ensure their effectiveness in various environments and conditions. Both artificial intelligence and machine learning are also expected to contribute to the rapid growth of the CRISPR system and parasite detection [
185,
186,
187,
188,
189]. Academic institutions conduct research, industries engage in industrial production, and governments and organizations (e.g., WHO) allocate resources, invest, and establish ethical guidelines to ensure universal access to CRISPR/Cas12a diagnostics in low-resource settings where parasitic diseases are most prevalent.

 ,
, 



{kind=link}
{kind=link}
{kind=link}