Cotton is a highly significant fiber crop that is extensively cultivated worldwide. Over the years, there has been a strong focus on enhancing the quality of cotton fibers, which has emerged as a prominent area of research. Cotton breeders have been actively engaged in develo** and cultivating germplasm resources that not only yield high quantities but also possess exceptional fiber quality [
1,
2]. Cotton fiber development is a complex process that occurs in four consecutive and overlap** stages. These stages include fiber initiation, elongation, thickening of the secondary cell wall, and maturation. Fiber initiation occurs from three days before flowering to three days post anthesis (DPA). Elongation occurs from 3 to 16 DPA. During this period, the cotton fibers undergo significant elongation as they grow in length. The secondary cell wall (SCW) stage typically spans from 16 to 40 DPA. During this phase, the cotton fibers undergo SCW deposition, resulting in increased fiber diameter and strength. The thickening of the SCW is an important process in enhancing the overall quality and strength of cotton fibers. The final stage of cotton fiber development is maturation, which occurs from 40 to 50 DPA [
3]. The number of fibroblast-differentiated protrusions affects the number of mature fibers to a certain extent, and the progression of rapid elongation through primary wall synthesis determines the length of the mature fibers. Cellulose is deposited in the SCW, and the cell wall is thickened during the thickening period of the SCW stage [
4,
5,
6]. Scientists are working to elucidate the key regulatory mechanisms involved in fiber development, including fibroblast differentiation, cell wall synthesis, and cellulose biosynthesis [
7,
8,
9]. Cotton fibers develop from ovule epidermal cells, and improving cotton fiber quality has long been a research hotspot. The creation and breeding of germplasm resources with high yields and excellent fiber quality has also become a long-term research direction and goal for cotton breeders [
1,
2]. Studying the regulatory mechanism of cotton fiber cell elongation and secondary wall development is highly important for improving the cotton yield and fiber quality.
In recent years, there has been significant progress in sequencing technologies, leading to their widespread use and continuous optimization. This has resulted in a growing body of evidence highlighting the crucial role of RNA transcriptional regulation in plant growth and development [
10]. Transcriptome sequencing (RNA-seq), one of the most commonly used second-generation high-throughput sequencing methods, has been widely used in the study of cotton fibers [
11,
12]. Through transcriptomics, many key regulatory pathways and gene expression patterns associated with fiber development have been revealed [
13,
14,
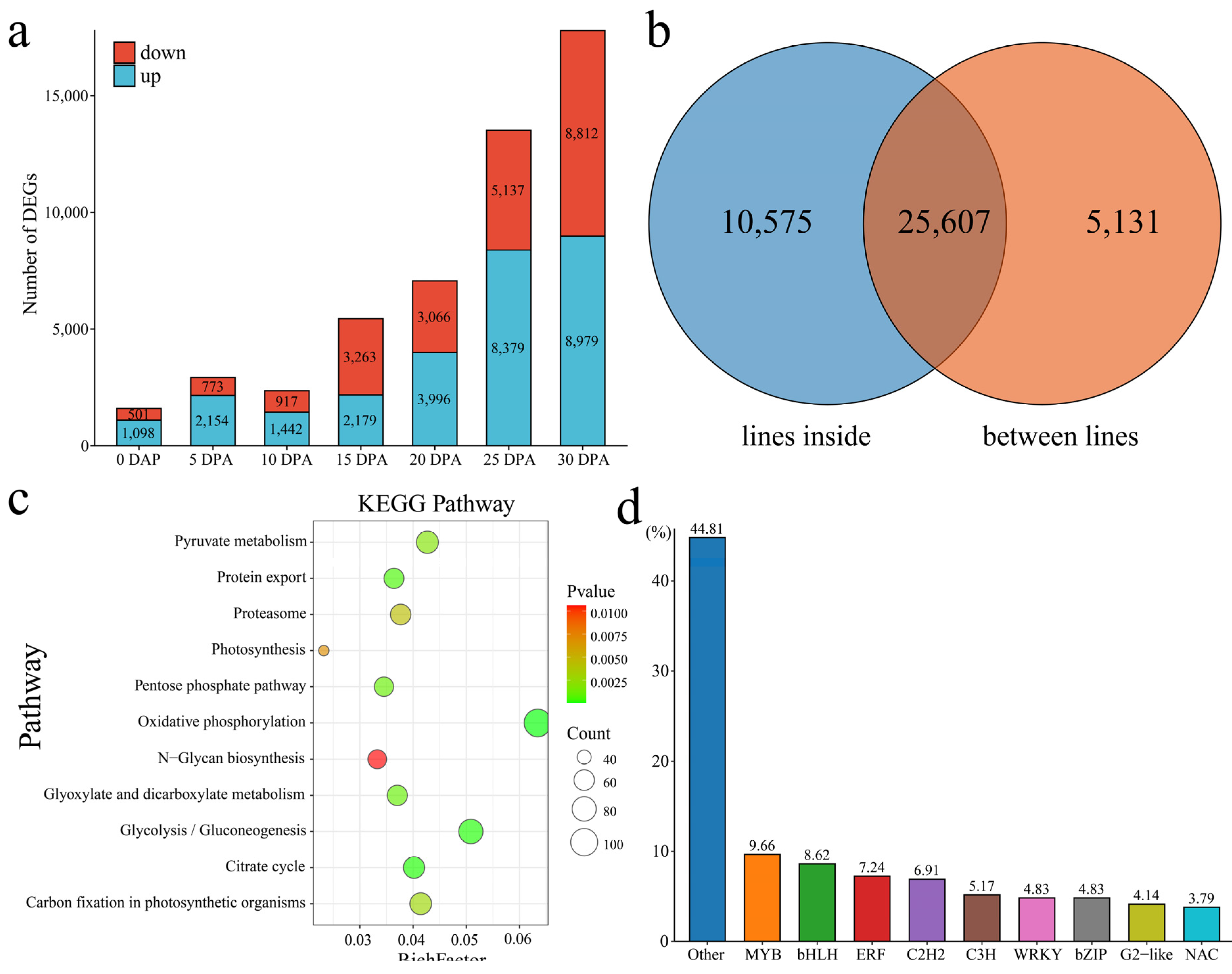
15]. These findings have helped identify the key genes that control fiber development and have provided important insights for improving cotton fibers. Through RNA-seq of 0–35 DPA fibers of PimaS-7 and 5917, 4 candidate genes related to fiber strength were identified [
16]. RNA-seq analysis at different stages of fiber development (7, 14, and 26 DPA) in the Coker 312 cotton variety allowed the identification of transcription factors and functional genes associated with this process. These genes encode proteins involved in various functional and metabolic pathways, including those involved in catalytic activity, carbohydrate metabolism, cell membrane and organelle functions, and signal transduction [
17]. Analysis of the RNA-seq data at 10 and 20 DPA from fibers from 4 wild cotton species and 5 domesticated cotton species showed that wild cotton plants allocate more resources to the stress response pathway and that acclimation may lead to a reprogramming of resource allocation in the direction of increasing fiber growth via regulation of the stress response network [
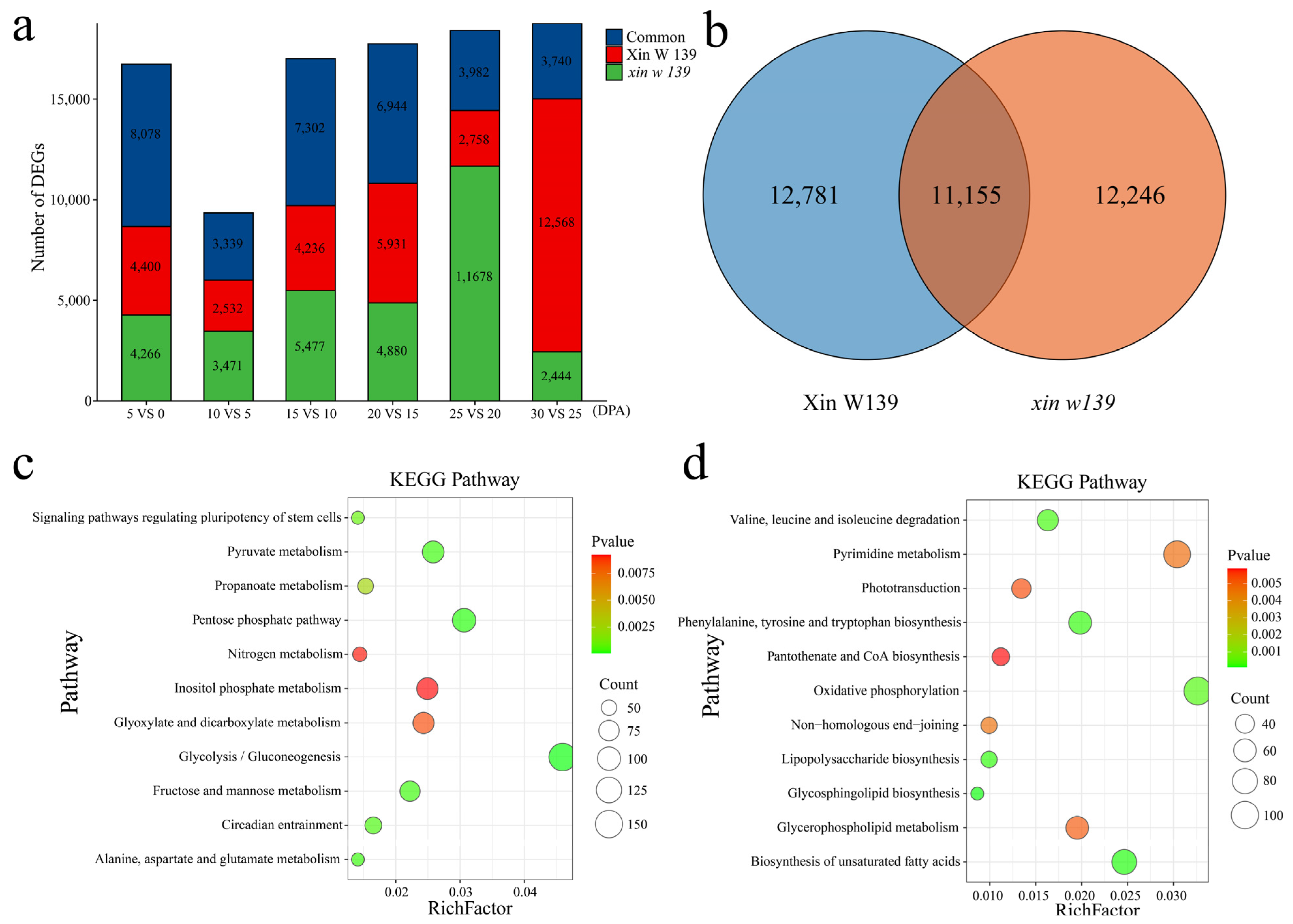
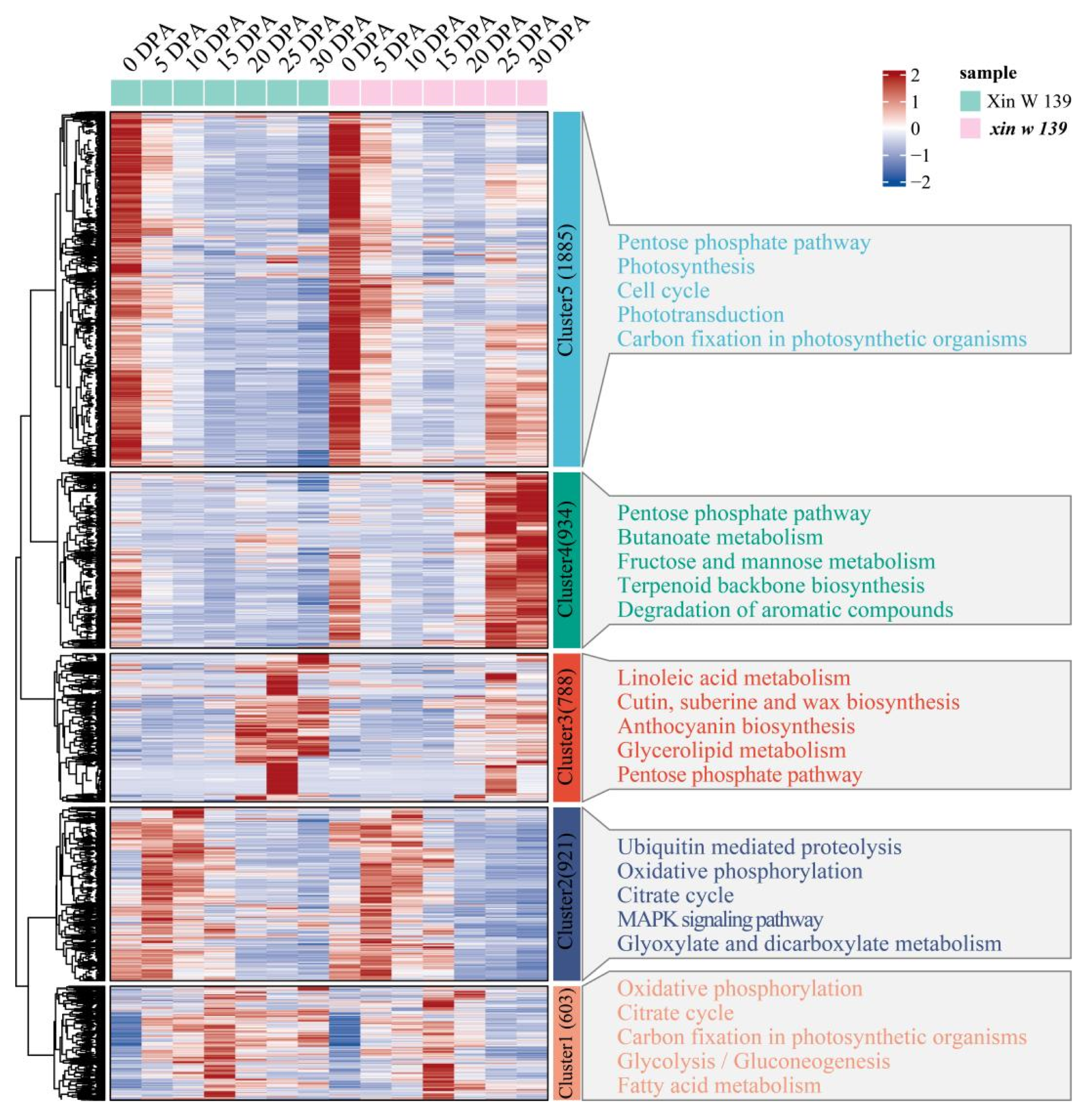
18]. Through RNA-seq analysis of the offspring of the ** stages. Most related studies have been based on cultivars that are promoted in production or based on lines that are used in breeding; especially, there are few reports on natural mutants [
25,
26,
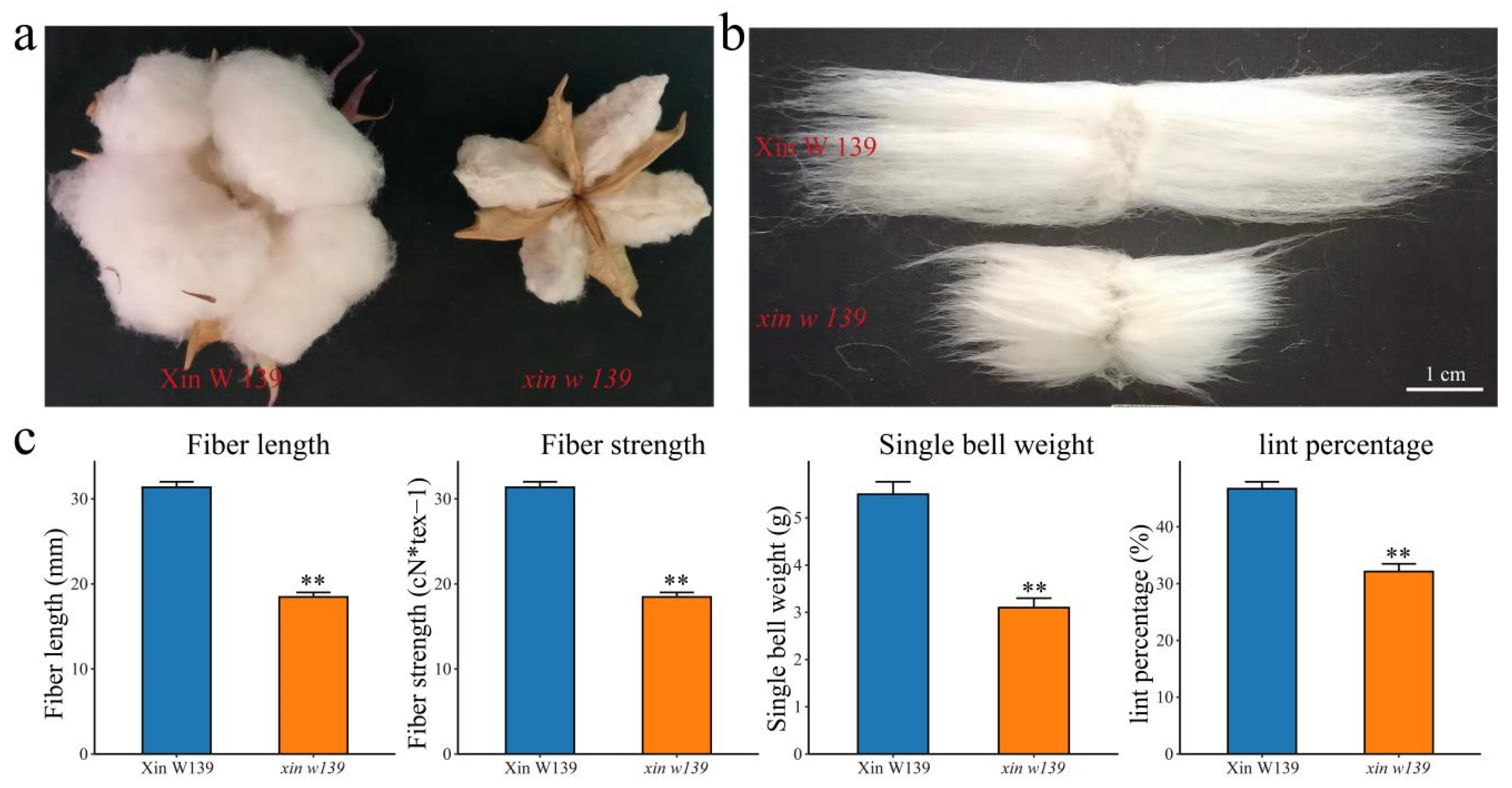
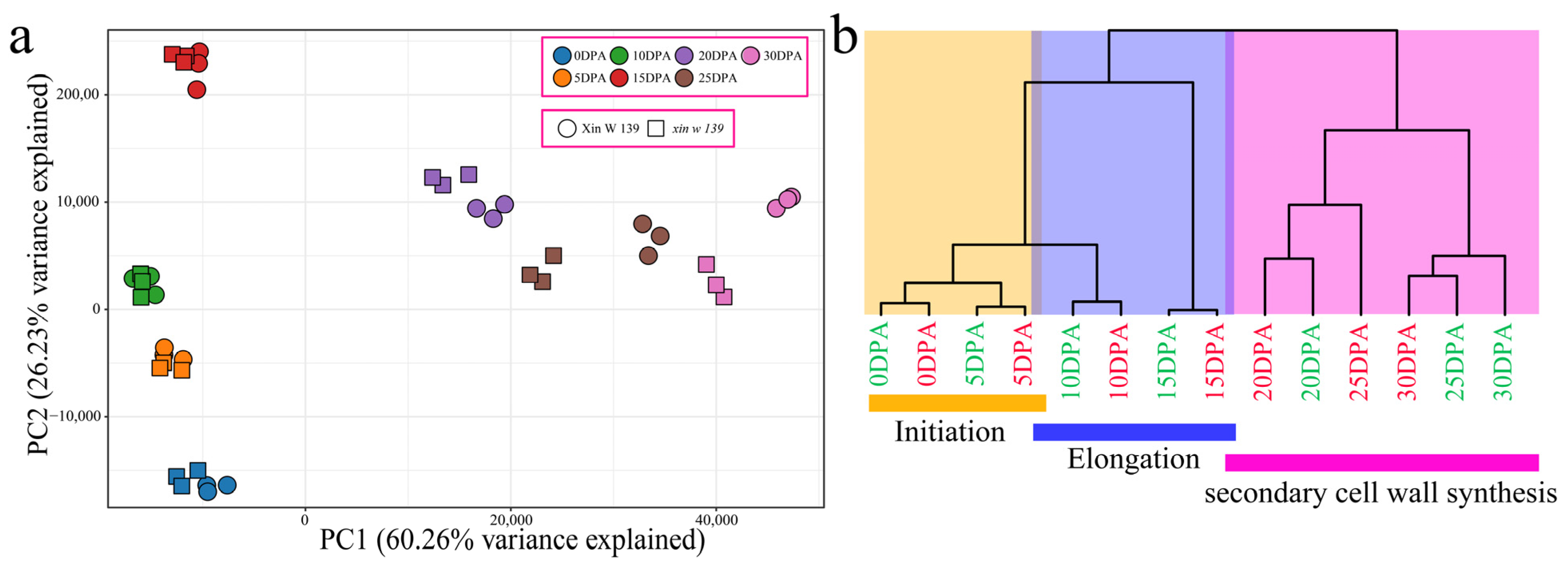
27]. Here, we performed phenotypic evaluation and comparative transcriptomic studies at seven time points during cotton fiber development in immature fiber mutant (
xin w 139) and wild-type (** of the
im mutant revealed that the gene encoding the PPR protein (
Gh_A03G0489) is related to the immature fiber phenotype of the
im mutant [
37]. Although the expression levels of
Gh_A03G0489 were significantly different between TM-1 and IM during the SCW period, the expression levels were relatively low [
37]. The PPR gene family is the largest gene family identified to date [
38]. They mainly act on mitochondria or chloroplasts to regulate organelle genes at the post-transcriptional level, thereby affecting plant growth and development [
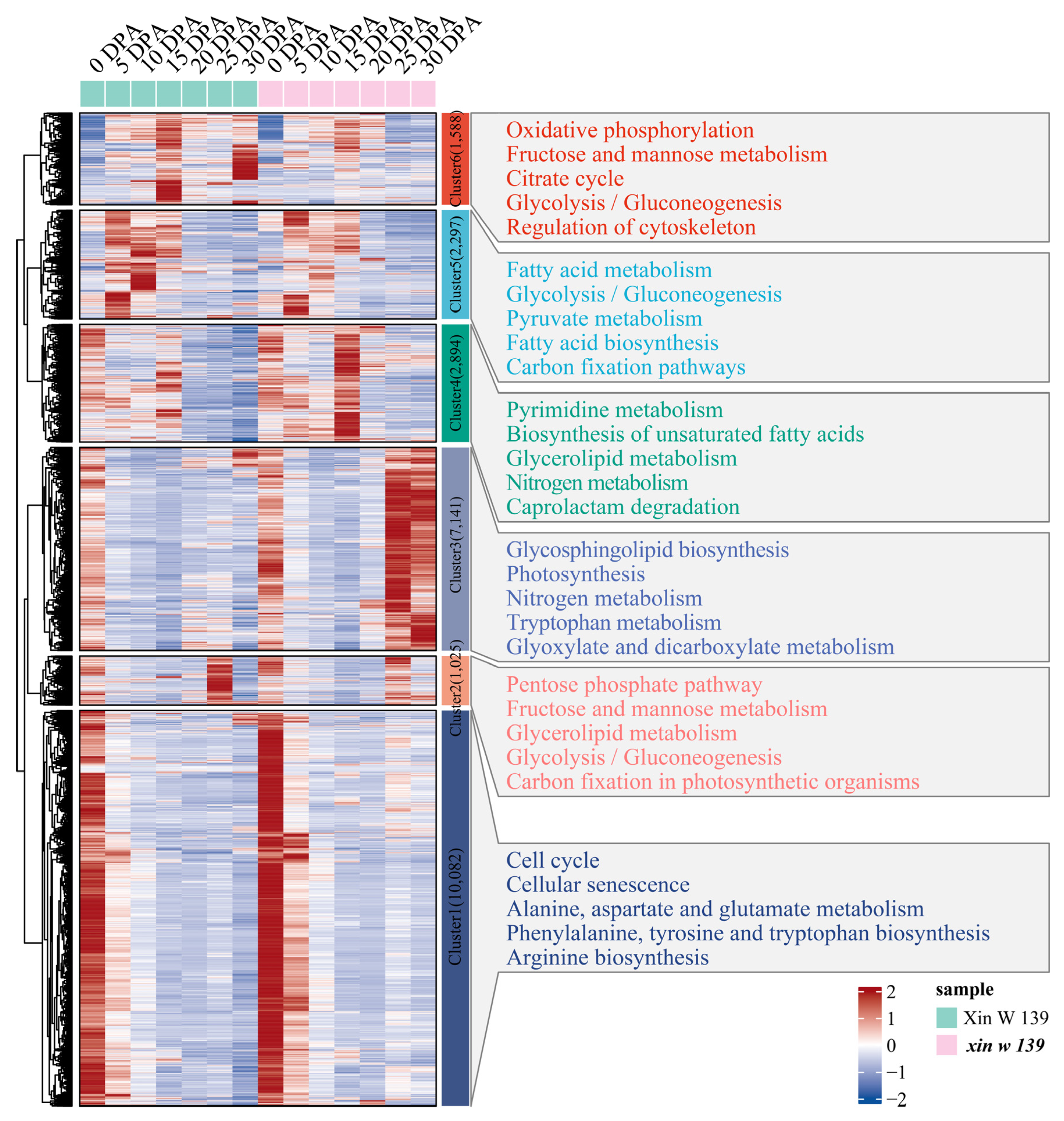
38]. We identified 523 genes encoding PPR proteins among the DEGs, and the expression levels in both lines were generally low (FPKM < 2). Notably, we found that two genes encoding PPR proteins (
GH_D11G0868 and
GH_A05G0653) exhibited a greater than four-fold change in expression during the SCW period (
Table S3). Mei et al. reported that
GH_A05G0653 was expressed mainly in the A subgenome of 4
G. hirsutum varieties at 20 DPA but showed no difference among the 3
G. barbadense varieties [
39]. We also found that the expression of
GH_A05G0653 in
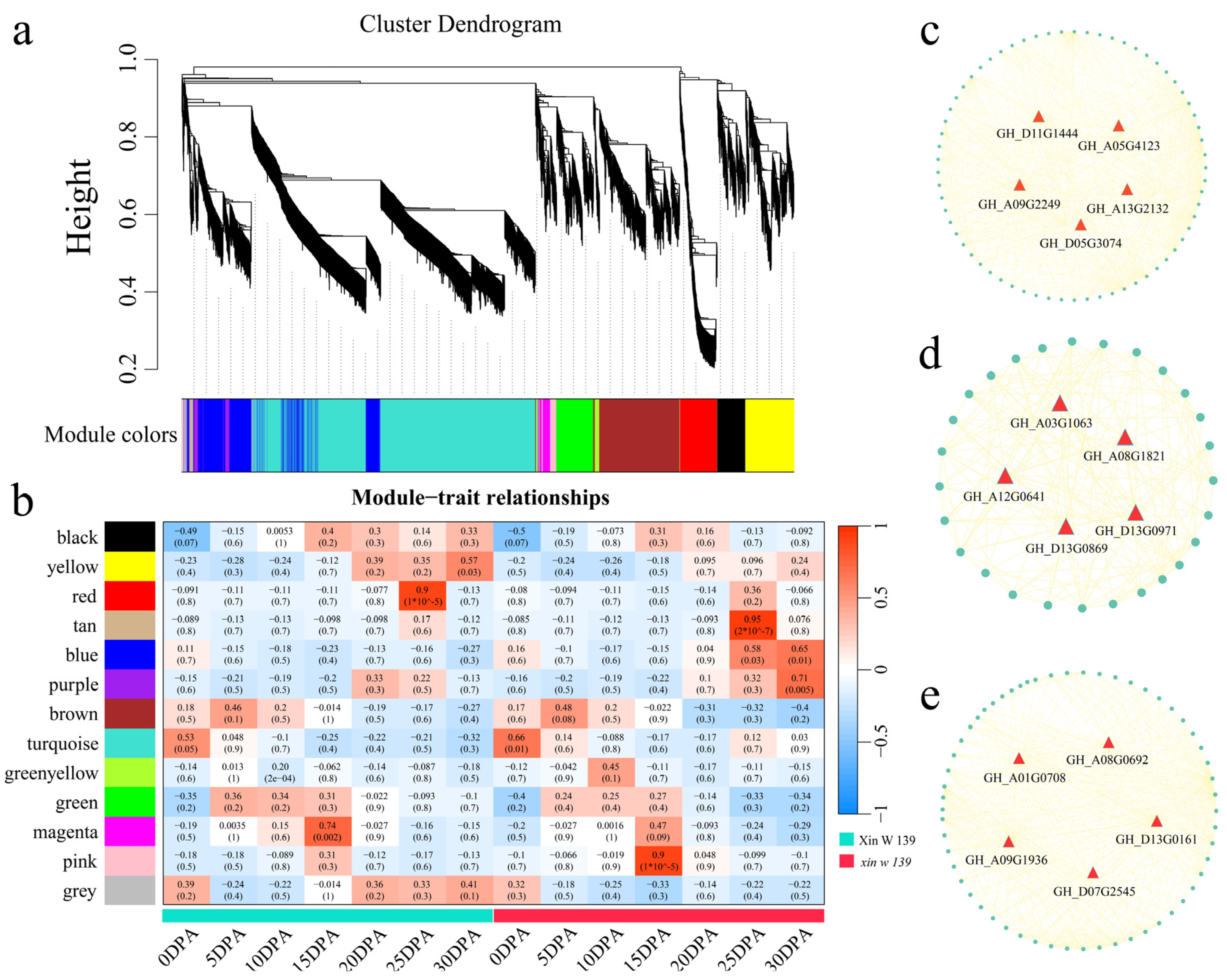
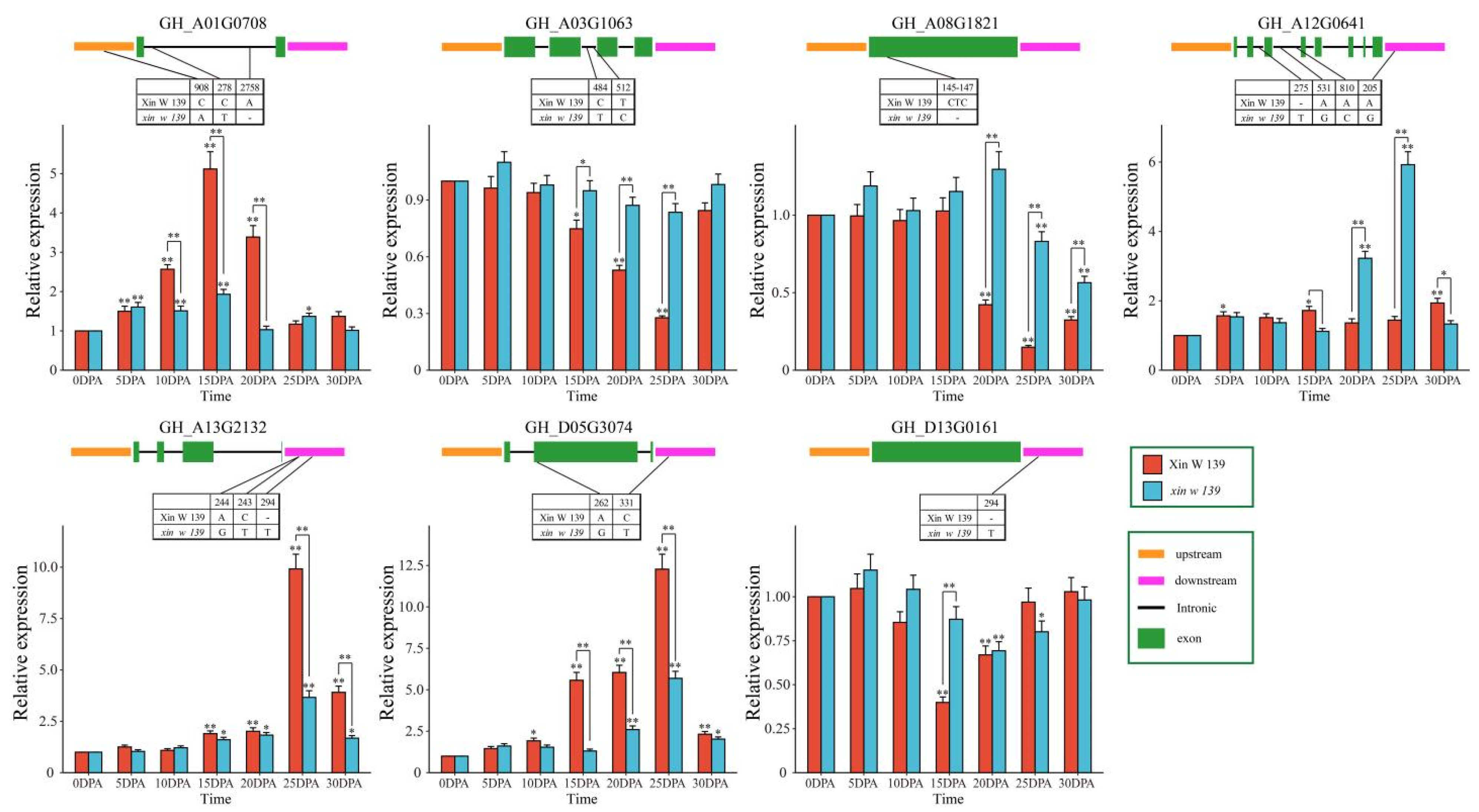
xin w 139 at 20 DPA was greater than that in ** signaling pathway, the pentose phosphate pathway. The pentose phosphate pathway produces NADPH, and NADPH is involved in fatty acid synthesis [
47]. The expression of the ethylene synthesis gene
GhACO1 was upregulated after the addition of very long-chain fatty acids (VLCFAs) in vitro, which increased the ethylene content in cotton fibers, thereby promoting the elongation of cotton fibers [
48]. A recent study showed that BRs regulate the synthesis of
GhKCS-mediated VLCFAs through
GhBES1, promoting fiber elongation [
49]. We found that the DEGs in
xin w 139 were annotated to the biosynthesis of the unsaturated fatty acids pathway, suggesting that the difference in fiber length between the two lines may be regulated by the expression of genes involved in the metabolic pathways of unsaturated fatty acids. VLCFAs, via enhanced UDP-l-rhamnose and UDP-d-galacturonic acid biosynthesis, can also increase the elongation of cotton fibers, which may also regulate fiber elongation through the sphingolipid biosynthesis pathway [
1]. However, the contribution of VLCFAs to fiber strength has not been reported. Due to the significant difference in fiber length and fiber strength between the two lines, we suspect that the pentose phosphate pathway contributes to both the lightness and length of the fibers; this hypothesis still needs to be verified, but our results can provide important information for subsequent studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







