3.1. Geometric Structure and Stability of TM@MoSSe
The structures of adsorbed TM single atoms on MoSSe are shown in
Figure 1a,b. Due to the special asymmetric structure of two-dimensional Janus material MoSSe, comprehensive consideration must be taken into account when considering the adsorption sites of transition metal single atoms on the surface of the material, and then stability analysis and performance research. Three different adsorption sites were considered for the S atomic layer on MoSSe, namely the Hollow site, the S atomic site, and the Mo atomic site. Similarly, on the Se atomic layer side, three different adsorption sites of transition metals on MoSSe were also considered, namely the Hollow site, Se atomic site, and Mo atomic site.
In order to find the most suitable SACs for OERs and HERs, the stability of different TM atom loads such as Fe, Co, Ni, Ru, Rh, Pd, Ag, Ir, Pt and Au on MoSSe surface was investigated. Firstly, the adsorption energies of different TM atoms on MoSSe were calculated to evaluate the stability and determine the geometric structures of different SACs. As shown in
Figure 1c,d, the adsorption energy calculation results show that Co, Ni, Ru, Rh, Pd, Ir, Pt, and Au had the most negative adsorption energy at the Mo site, followed by the Hollow site, and the most positive adsorption energy at the S/Se site on base MoSSe. The adsorption energy of a Ag single atom at Mo site and Hollow site was similar, and the adsorption energy value at the S/Se site was the most positive and the adsorption was the most unstable. Compared with the adsorption of other TM single atoms on MoSSe, the adsorption of Ag single atoms was weak (the adsorption energy was −0.57~−0.91 eV), and Ag single atoms cannot form stable single-atom catalysts on the surface from an energy point of view. In conclusion, TM (Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt, and Au) single atoms can stably adsorb on Mo atomic sites of both the Se side and S side on MoSSe, and further comparison of adsorption energies showed that the adsorption of TM single atoms on the S side of MoSSe was more stable. Therefore, the stable model of TM@MoSSe was preliminarily determined as TM single atoms supported on Mo sites in the S layer on MoSSe.
It is worth noting that the dissolution potential (U
diss) is also very important for the stability evaluation of electrocatalytic materials [
39]. More negative adsorption energy means that TM atoms adsorbed on MoSSe are thermodynamically stable, while the corrected U
diss indicates that TM atoms are more electrochemically stable. The stability of the material in the electrochemical reaction environment can be indicated only when both of them reach a certain standard. As shown in
Figure 1e,f, the calculation results of the dissolution potential show that Fe Co, Ni, Ru, Rh, Pd, Ir, Pt, and Au had the largest dissolution potential at the Mo site on the MoSSe, followed by the Hollow site, and the smallest at the S/Se site. The dissolution potential of Fe and Ag at the Mo site and Hollow site was similar, and the solubility potential at the S/Se site was the least, which means that it was the most unstable. The calculation result of dissolution potential is consistent with the adsorption energy, that is, the stability model of TM@MoSSe is TM (Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt, Au) loaded on the Mo atomic site in the S layer of MoSSe.
Thermodynamic calculation alone is not enough to describe the stability of materials at practical application temperature. On the basis of energy calculation, the stability of materials is further determined by AIMD calculation. Firstly, the TM@MoSSe (TM = Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt, Au) samples were assigned an initial temperature of 100 K, and then heated up to the desired temperature 500 K over 3 Ps. During the simulation process, the position of Au atoms on the Au@MoSSe surface changed with temperature, indicating that Au atoms cannot be anchored on the surface of MoSSe to form single atomic catalysts, as shown in
Figure S1. For other TM@MoSSe (TM = Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt) materials, the structure of a single atom anchored on MoSSe surface was very stable from 100 to 500 K.
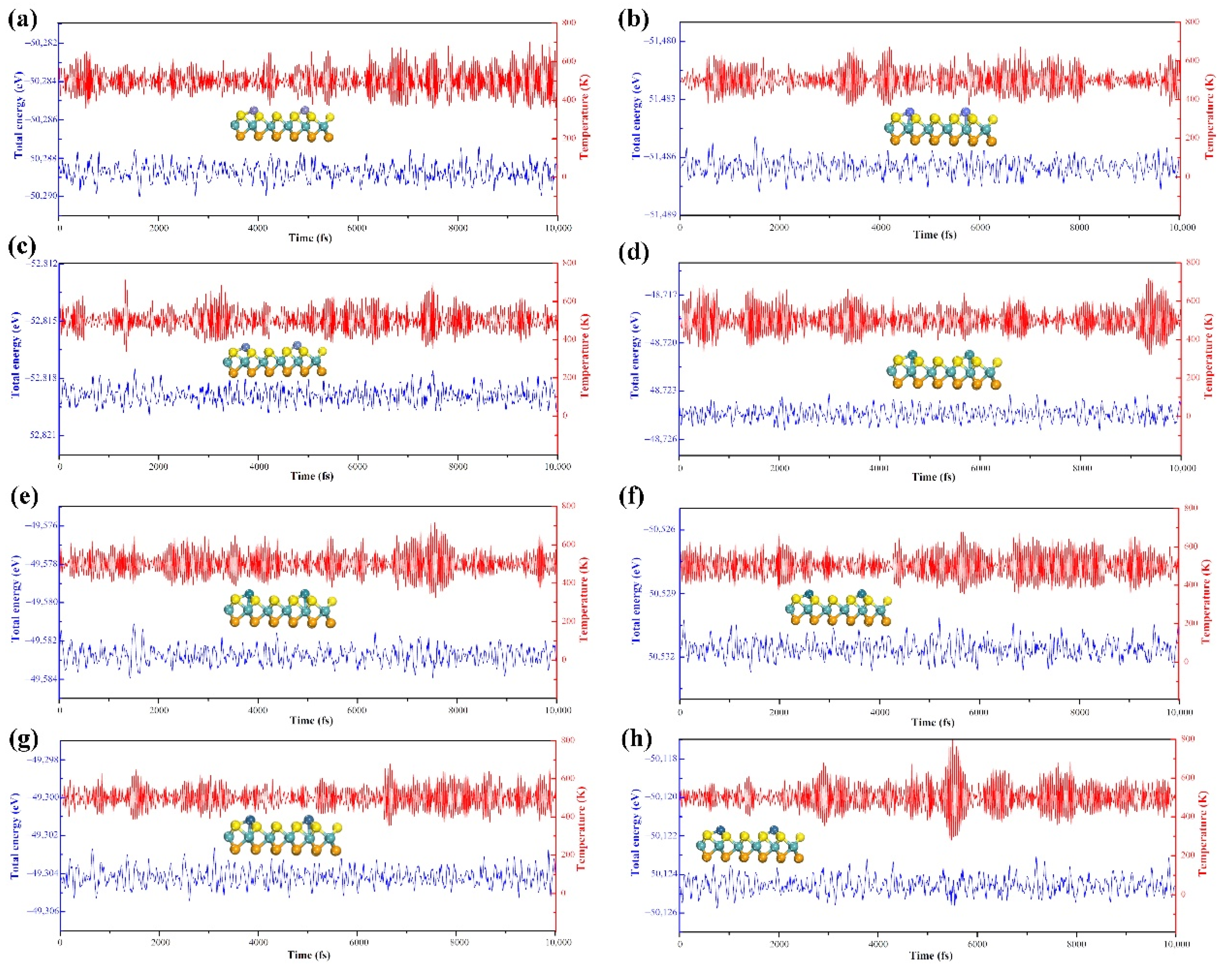
In addition, we further performed an AIMD simulation for 10 ps. As shown in
Figure 2, the total energy and the temperature oscillated near the equilibrium state. Meanwhile, the structure of the single atom anchored on MoSSe surface still had no significant change (the TM@MoSSe trajectories of AIMD simulation shown in
Figure S2), indicating the high stability of the TM@MoSSe material. Therefore, we can conclude that the single TM (Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt) atom anchored on MoSSe surface is stable and the next discussion of catalytic performance is based on the screening in this section.
3.2. HER and OER Catalytic Activity
The original unmodified surface of MoSSe is inert and not an excellent catalyst for HERs and OERs. After screening the stable TM@MoSSe structure, the single atom anchored on the surface was used as the catalytic active site to explore their HER and OER performance. To evaluate the HER activity of TM@MoSSe, we calculated the Gibbs free energy (ΔG
H) of the hydrogenation process based on the method proposed by Norskov et al. [
40,
41], in which the free energy of H
++ e was replaced by half the chemical potential of a hydrogen molecule. The absolute value of ΔG
H signifies the energy barrier to be overcome for HERs, a positive value of ΔG
H indicating that the binding strength between the H atom and catalyst is strong and that it is difficult to desorb, while a negative value of ΔG
H indicating that the interaction between H and the catalyst is weak and that it is difficult to adsorb.
As shown in
Figure 3c, all TM@MoSSe catalysts exhibited better HER activity than the original MoSSe materials, indicating that anchoring a single transition metal atom on MoSSe is a feasible method to improve the catalytic performance of MoSSe materials. It is worth noting that the values of Fe@MoSSe, Ru@MoSSe, Rh@MoSSe, and Pt@MoSSe ΔG
H were 0.08 eV, −0.05 eV, 0.03 eV, and −0.06 eV, which are close to 0. As is well-known, the high cost and low storage of these precious metals (Ru, Rh, Pt) seriously limit their practical application in energy conversion and storage. The Fe element, with low cost and high reserves, is one of the candidate materials for catalysts that are expected to replace precious metals. Furthermore, it is reported that a variety of Fe single-atom catalysts have been successfully synthesized and used for HER electrocatalysts, which can ensure good stability and activity in acidic [
42] or alkaline [
43] electrolyte media. Therefore, the Fe@MoSSe can be used as a potential HER electrocatalytic material.
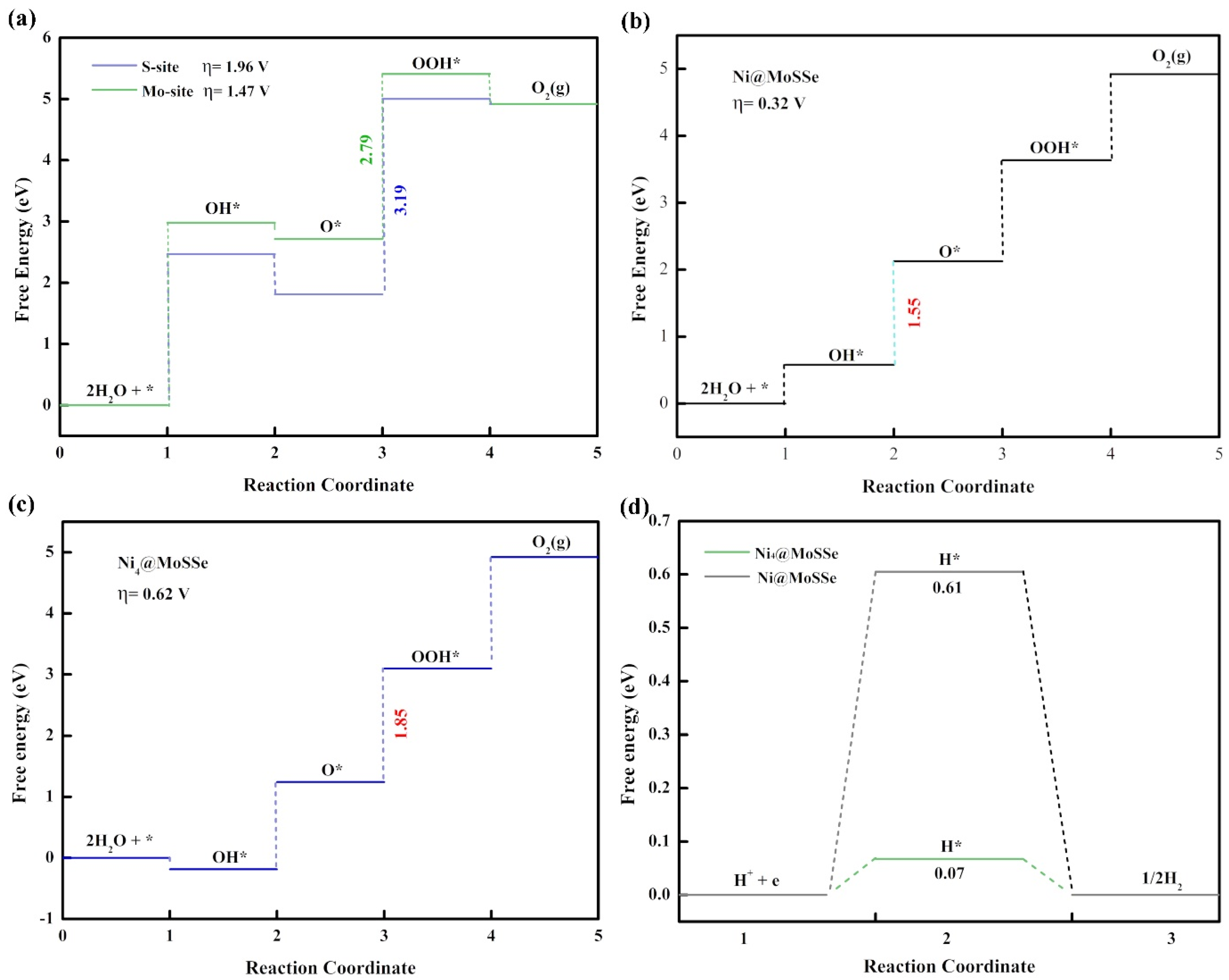
The OER performance of the original MoSSe was calculated by considering two different reactive active sites (i.e., the Mo atom site on the S side and the Mo atom site on the Se side). As shown in
Figure 4a, the calculated Gibbs free energy of the four-step OER reaction process showed that the OER overpotential was 1.96 V at the Mo atom site on the S side and 1.47 V at the Mo atom site on the Se side. In the third step, the energy required for the reaction of O* with OH* to generate the intermediate of HOO* was the largest. This step is a key step to determine the overpotential of the electrocatalytic oxygen evolution reaction on MoSSe. The large overpotential indicated that the catalysis activity of MoSSe needs to be improved, and MoSSe without any surface modification is not an excellent electrocatalytic material that can replace precious metals.
The OER reaction process includes four elementary steps, each of which is accompanied by proton and electron transfer. For the elementary step, the change of Gibbs free energy (ΔG) of the four step reactions can be calculated according to the first-principles calculation. The ΔG value of the largest of the four-steps will determine the overpotential of OER (η).
Figure 3a shows the OER overpotential calculation results of TM@MoSSe (TM = Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt) from left to right. The overpotential was 1.02 V, 0.79 V, 0.32 V, 1.16 V, 0.68 V, 0.55 V, 0.93 V and 0.55 V, respectively. It is worth noting that Ni@MoSSe had the smallest overpotential value (η = 0.32 V) during a series of TM@MoSSe, and the dissociation of OH* to O* is the key step to determining the OER overpotential, shown in
Figure 4b. The overpotential of Ni@MoSSe was 1.15 eV lower than that of the original MoSSe, and Ni@MoSSe has better catalytic performance than the typical OER catalyst IrO
2 (η = 0.42 V). In previous experimental studies [
44], IrOx was found to be the only stable catalyst in acidic media compared with non-precious metal catalysts. In addition, although IrOx is unstable, no non-precious metal catalysts had the same activity as IrOx under the same conditions. In this work, the calculation results show that it is possible to break this relationship by preparing non-precious metal single-atom catalysts. The dissolution potential and AIMD simulation indicated that the non-noble metal single-atom anchored MoSSe catalyst has strong stability. As is well-known, single-atom catalysts improve the utilization of metals and increase the number of active sites. By anchoring different metals on the surface of MoSSe, the d-band center of the catalyst is further adjusted to reduce the overpotential of OERs.
All free energy diagrams for the OER of the TM@MoSSe at an electrode potential of U = 0 V are shown in
Figure S3.
Interestingly, as shown in
Figure S4, when the Gibbs free energy of TM@MoSSe changes, it is found that the adsorption of Gibbs free energy of intermediate O* is significantly correlated with the number of d electrons adsorbed on transition metal atoms. In the same period, as the number of transition metal d electrons increases, the ΔG value of the second step reaction also increases. The above phenomena indicate that the number of d electrons of TM atoms will affect the energy change of the OER process, which can provide some guidance for the follow-up study on the source of OER activity of TM@MoSSe. In addition, under the experimental conditions, some single-atom catalysts formed clusters on the surface, thus affecting the catalytic activity of the catalysts. Therefore, a Ni4 cluster model was established on the MoSSe surface to investigate the effect of clusters on the catalytic activity. Furthermore, the results show that Ni@MoSSe can maintain good OER and HER activities even if isolated Ni atoms are clustered into small clusters, as shown in
Figure 4c,d. See the
Supplementary File for a detailed discussion.
3.3. The Origin of Catalytic Activity of TM@MoSSe
By anchoring transition metal single atoms on the surface, the catalytic performance of the original MoSSe was greatly improved. In order to understand the source of excellent catalytic activity, the charge of TM@MoSSe was calculated and analyzed. As shown in
Table 1, the series TM@MoSSe Bader charge can be used to quantitatively calculate the charge of the system, and the number of electrons around the atom can be obtained, so as to approximate the valence of the atom. The data analysis shows that after the adsorption of transition metal single atoms, the charge of the material is transferred to different degrees, in which Se and S atoms obtain electrons, and the S atom obtains more charges than Se. The main atoms that lose electrons are Mo atoms and doped transition metals. This shows that after the transition metal single atom is anchored, the charge in the MoSSe system is redistributed, the electrons in the Se layer are transferred to the S layer and the transition metal atoms on the surface.. The change of the electronic structure is the main reason for the difference in material performance, and the intervention of the transition metal single atom effectively regulates the electronic structure of the material itself.
The good conductivity of the catalyst itself can greatly eliminate the energy barrier at the catalyst–electrolyte interface and the electrode interface in the electrocatalytic reaction, and ensure high-energy conversion efficiency. The density of states of the original MoSSe was calculated. As shown in
Figure S5, the original MoSSe is a semiconductor material with a band gap of 1.62 eV. From the density-of-states diagram analysis, it can be seen that, compared with non-metallic elements, such as S and Se, the d-electronic states of Mo atoms contribute more to the electronic states near the Fermi level.
The OER performance of Ni@MoSSe is very prominent in a series of TM@MoSSe. In order to explore the essential reason for the catalytic activity of Ni@MoSSe, the DOS was analyzed. As shown in
Figure S6, the band gap of the material decreased to a certain extent after the single atom Ni was loaded on the surface of MoSSe, indicating that the conductivity of MoSSe was improved to a certain extent after the modification of single atom Ni. At the same time, the other TM@MoSSe also had the same tendency, indicating that transition metal loading can improve the catalytic activity of MoSSe material.
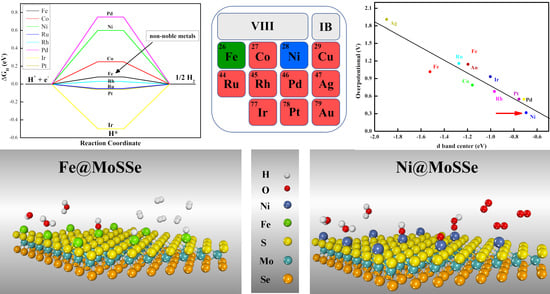
For metal catalysts, the active element composition, chemical state, size, and the crystal structure of all sorts of complicated factors, such as extremely subtle structure change, can bring huge performance change. Therefore, it is very difficult to adjust the performance of the metal catalyst. According to the d-band center theory, the d-band center of a series of TM@MoSSe metals was explored and fitted with overpotential. It was found that there was a good linear relationship between the d-band center and material OER overpotential (R
2 = 0.85), as shown in
Figure 5. In the TM@MoSSe system, the stability and activity of the catalysts are improved and the OER overpotential of the materials is decreased with an increase of the binding strength between the transition metals and the MoSSe surface.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}